

3.2.Основные принципы описания молекул в квантовой химии и метод валентных связей (мвс).
3.2.1. Основные принципы описания молекул в квантовой химии
Теоретическое обоснование энергетической целесообразности образования химической связи между изолированными атомами стало возможным только после того, как были разработаны способы описания движения микрочастиц, т.е. после открытия законов квантовой механики. В основе данного обоснования лежат следующие принципы квантовомеханических расчётов:
1.Решение уравнения Шредингера с использованием приближённых волновых функций.
Уравнение Шредингера, описывающее поведение электрона в атоме водорода имеет вид:
▼2ψ + 8π2m/h2(E – V)ψ = 0 (1.3)
где ▼2– оператор Лапласа и, следовательно, ▼2ψ = ∂ψ/∂х2+ ∂ψ/∂y2+ ∂ψ/∂z2; Е – полная энергия системы,V– её потенциальная и (E–V) кинетическая энергия, соответственно.
Это уравнение может быть представлено в виде:
{ (- h2/8π2m)▼2 + V}ψ = Eψ или сокращённо: Ĥψ = Eψ (2.3)
где Ĥ– оператор Гамильтона, определяющий операцию или последовательность операций, производимых над функцией ψ.
Если умножить обе части последнего уравнения на ψ и проинтегрировать по всему пространству ( от -∞ до +∞ по каждой из координат), то можно получить выражение, описывающее полную энергию системы:
∫ ψĤψ dv / ∫ ψ2dv = E (3.3 )
Полученное соотношение является одним из основных уравнений квантовой химии.
Так как точный вид ψ-функций известен только для одноэлектронных систем, в квантовой химии используют их приближённые выражения. При этом приближённые значения функций ψ определяют с помощью вариационного метода. Оптимальную функцию в рамках этого метода находят путём последовательного приближения, постулируя, что ближе к истинному выражению будет тот вид ψ, подстановка которого в уравнение (3.3), позволит получить минимальное значение Е. В подавляющем большинстве случаев функцию ψ выражают в виде суммы:
Ψ = с1φ1 + с2φ2 + с3φ3 + ………. + сnφn (4.3)
где φ – функция зависящая от координат,с- коэффициенты
Коэффициенты сi подбираются с учётом основного принципа рассматриваемого метода – принципа минимальной энергии системы:
∂Е/∂с1 = 0; ∂Е/∂с2 = 0; ∂Е/∂с3 = 0; …………… ∂Е/∂сn = 0. (5.3 )
2. Расчет энергии молекул и молекулярных ионов.
Изменение энергии в системе состоящей из двух ядер показано на рис.7.3 При расчете энергии в подобных случаях за нулевую принимается энергия системы, в которой невозбуждённые атомы удалены друг от друга на бесконечно большое расстояние. При сближении атомов водорода между ними возникают силы притяжения, что приводит к снижению энергии системы в целом. Изменение энергии системы наблюдается то тех пор, пока силы отталкивания (резко возрастающие при сближении ядер атомов на расстояние меньше ¾ суммы радиусов атомов) не скомпенсируют силы притяжения. Таким образом, энергия системы складывается из суммарной полной энергии электронов и потенциальной энергии взаимодействия ядер. В связи с этим зависимость энергии системы от расстояния между центрами взаимодействующих атомов имеет минимум, который характеризует наиболее стабильное состояние бинарной молекулы. В невозбуждённой молекуле при Т > 0оК ядра атомов совершают колебания относительно равновесного расстояния re (рис.7.3), что увеличивает энергию системы на некоторую величину, которую обозначим через ε0. Тогда экспериментально найденная энергия связи (количество энергии выделяющейся при образовании одной связи данного типа в рассматриваемой молекуле) определяется соотношением Есвязи = Е min - ε0.
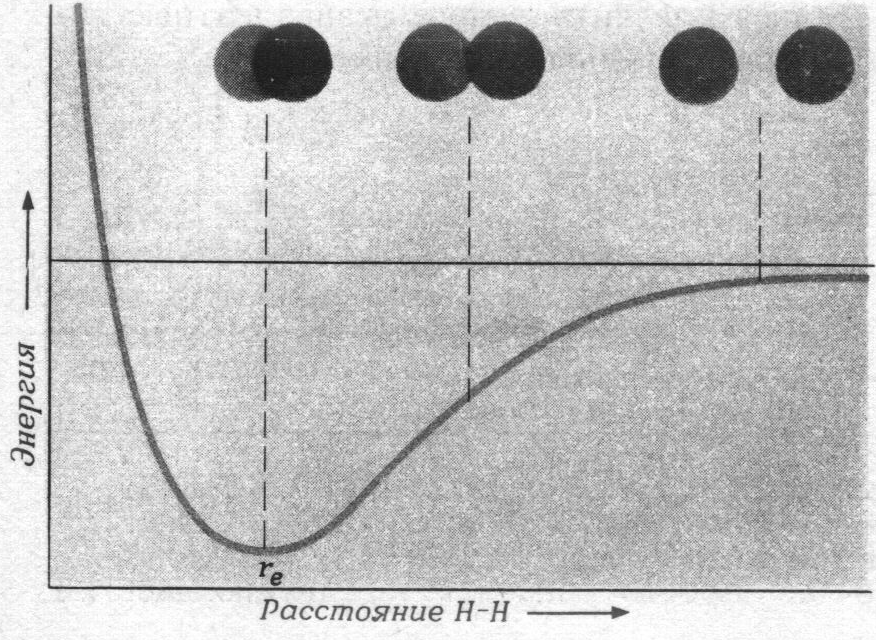
Рис.7.3. Теоретическая кривая изменения энергии связи в молекуле Н2 в зависимости от расстояния между ядрами атомов
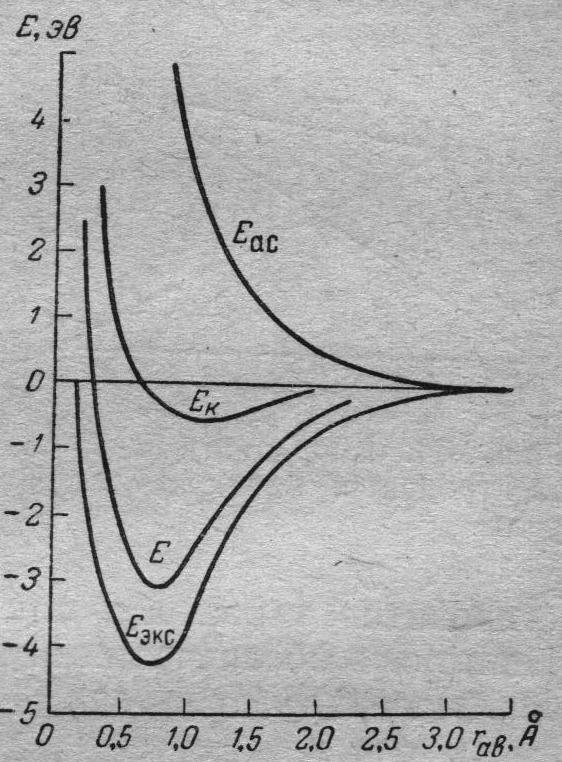
Рис. 8.3. Экспериментальная и теоретические кривые зависимости энергии в молекуле Н2 от расстояния между ядрами атомов. Ек – расчёт на основе модели кулоновского взаимодействия (модель Льюиса); Е и Еас. –рассчитаны с использованием симметричной и антисимметричной волновых функций, соответственно; Еэкс. – экспериментальные данные.
Как правило, в литературе при описании молекулярного вещества приводятся значения энергии одного моля рассматриваемых связей и значения re. Экспериментальные кривые (рис.8.3 кривая Еэкс.) определяют, используя данные молекулярных спектров.
Как отмечалось выше, квантовомеханические расчёты энергии молекулярных систем для различных значений (r) проводят с использованием уравнения (3.3). Критерием правильности такого расчёта является степень совпадения теоретической и экспериментальной кривых Е = f(r) в рассматриваемой системе.