Клаузиус назвал эту функцию энтропией (от греч. εντροπια – поворот, разворот), поскольку она характеризует степень обратимости термодинамических процессов.
Неравенство Клаузиуса в дифференциальной форме (21.19) было получено нами в лекции 19 из статистических представлений и больцмановского определения энтропии. Там же были сформулированы основные свойства энтропии, в том числе принцип возрастания энтропии в изолированной системе. Из него и вытекает второе начало термодинамики, которое мы теперь, следуя Больцману, можем сформулировать таким образом:
В изолированной системе все тепловые явления наиболее вероятно протекают в направлении, приводящем к возрастанию энтропии.
Таким образом, все предшествующие формулировки второго начала термодинамики теперь получили статистическое обоснование. Самопроизвольный переход тепла от менее нагретого тела к более нагретому, полное превращение теплоты в работу и уменьшение энтропии при необратимых процессах в изолированной системе – все эти явления нарушают тенденцию движения системы в сторону термодинамического равновесия. Однако для малых количеств передаваемой теплоты и для систем из небольшого количества частиц эти процессы не абсолютно запрещены, а всего лишь маловероятны. В больших системах они могут наблюдаться в виде флуктуаций – случайных отклонений от средних значений наблюдаемых величин.
§ 5. Третье начало термодинамики
Одним из свойств энтропии, вытекающих из неравенства Клаузиуса (см. § 4 лекции 19), является равенство энтропии любой системы нулю при температуре, равной абсолютному нулю:
Это утверждение называется теоремой Нернста в честь немецкого физико-химика В. Нернста, высказавшего его в 1906 г. Часто теорему Нернста называют третьим законом термодинамики:
При температуре, стремящейся к абсолютному нулю, энтропия любой термодинамической системы стремится к нулю.
Лекция 22
6.4. Фазовые состояния, переходы и равновесия
§ 1. Основные понятия
Под фазой в термодинамике понимается однородная (гомогенная) по химическому составу и физическим свойствам часть термодинамической системы, отделенная от других ее частей (фаз), имеющих иные свойства, границами раздела, на которых происходит изменение свойств.
Примерами различных термодинамических фаз одного и того же химического вещества – воды – являются жидкая вода, лед и водяной пар. Они могут встречаться вместе. Например, весной поверхность водоема еще покрыта льдом, но он тает под лучами солнца, и в воздухе появляются пары воды. Однако было бы ошибкой отождествлять
агрегатные состояния вещества (твердое, жидкое и газообразное) с
его термодинамическими фазами. Понятие фазы гораздо шире, чем агрегатное состояние, и различных фаз у вещества может быть гораздо больше трех. Так, в твердом состоянии вещество может образовывать несколько различных по симметрии и физическим свойствам кристаллических модификаций. Это явление называется аллотропией или полиморфизмом. Хорошо известны различные полиморфные модификации углерода: алмаз, графит, карбин – нитевидные кристаллы, состоящие из параллельно ориентированных цепочек углеродных атомов. К ним следует добавить различные наноаллотропы углерода: фуллерены, нанотрубки, наноленты, графен и т. п. низкоразмерные кристаллические структуры. Жидкое состояние вещества также может быть представлено различными фазами. Например, существуют
жидкий гелий-I (обычная жидкость) и жидкий гелий-II (сверхтекучая жидкость).
Фазы подразделяют на однокомпонентные и многокомпонент-
ные (смеси, сплавы и т. д.). В состоянии термодинамического равновесия потоки частиц между различными фазами также уравновешены, вследствие чего массы контактирующих фаз остаются неизменными. Говорят, что в этом случае имеет место фазовое равновесие. В усл о- виях фазового равновесия, при отсутствии химических реакций в фазах, выполняется правило фаз:
где Ф – число равновесных фаз в термодинамической системе, к – число компонентов в этой системе, i – число степеней свободы системы.
Под числом степеней свободы системы в данном случае понимается число ее параметров состояния, которые могут изменяться, будучи связанными друг с другом каким-либо соотношением. Так, например, в однокомпонентной системе (к =1) равновесие дух фаз
p 
Рис. 22.1. Диаграмма фазовых состоя-
ний и равновесий простого вещества ( pT -диаграмма)
(Ф = 2) характеризуется только одной степенью свободы (i =1). Это означает, что при заданном объеме (V = const) изменение
температуры ведет к измен е- нию только одного из трех термодинамических параметров – давления (см. рис. 22.1). В случае же равновесия трех фаз, согласно (22.1), число степеней
свободы системы равно нулю. Отсюда следует, что на pT -диаграмме равновесие между объемом, давлением и температурой возможно только в одной точке. Ее называют тройной точкой. Линия равновесия жидкость – пар (газообразное состояние) заканчивается в критической точке К, где теряется различие между жидкостью и ее паром.
Перейдем теперь к фазовым переходам. Прежде всего, отметим, что они бывают двух видов.
Фазовыми переходами первого рода называются такие фа-
зовые превращения, при которых происходит выделение или поглощение некоторого количества теплоты.
Примерами таких переходов, в частности, являются переходы из одного агрегатного состояния вещества в другое: плавление  кристаллизация, испарение
кристаллизация, испарение  конденсация, возгонка или сублимация (переход вещества из твердого состояния в газообразное, минуя жидкую фазу)
конденсация, возгонка или сублимация (переход вещества из твердого состояния в газообразное, минуя жидкую фазу)  десублимация. При этом теплота поглощается, если при фазовом переходе происходит разрыв связей между молекулами и увеличение степени хаотичности их движения. Напротив, при образовании дополнительных связей между молекулами теплота выделяется.
десублимация. При этом теплота поглощается, если при фазовом переходе происходит разрыв связей между молекулами и увеличение степени хаотичности их движения. Напротив, при образовании дополнительных связей между молекулами теплота выделяется.
Фазовыми переходами второго рода называются такие фа-
зовые превращения, при которых теплота не выделяется и не поглощается, но имеет место скачкообразное изменение некоторых физических констант вещества (теплоемкости, вязкости, электропроводности, диэлектрической или магнитной проницаемости и т. п.).
Примерами фазовых переходов второго рода являются ферромагнетик 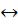 парамагнетик, сегнетоэлектрик
парамагнетик, сегнетоэлектрик  диэлектрик, переходы в сверхпроводящее и сверхтекучее состояния и обратно.
диэлектрик, переходы в сверхпроводящее и сверхтекучее состояния и обратно.
§ 2. Уравнение Клапейрона – Клаузиуса
Данное уравнение имеет отношение к фазовым переходам первого рода в однокомпонентной термодинамической системе в условиях двухфазного равновесия. Рассмотрим его вывод на примере фазового перехода жидкость  насыщенный пар.
насыщенный пар.
p 
dp
Рис. 22.2. К выводу уравнения Клапейрона - Клаузиуса
Пусть рабочим телом будет система жидкость – насыщенный пар. Осуществим над ней цикл Карно (рис. 22.2). Учтем, что на изотерме в этой двухфазной системе давление будет постоянным. Пусть удельная теплота фазового перехода равна λ . Переведем при температуре Т массу m вещества из жидкой фазы в паровую (процесс 1  2). Затем переведем систему адиабатически в бесконечно близкое состояние (процесс 2
2). Затем переведем систему адиабатически в бесконечно близкое состояние (процесс 2  3). Теплота испарения в этом процессе берется от самой жидкости, в результате чего ее температура понизится на величину dT . Это автоматически приведет к понижению давления пара на величину dp . Далее завершим цикл переходами 3
3). Теплота испарения в этом процессе берется от самой жидкости, в результате чего ее температура понизится на величину dT . Это автоматически приведет к понижению давления пара на величину dp . Далее завершим цикл переходами 3  4 и 4
4 и 4  1.
1.
Работа, совершаемая в данном цикле, равна его площади. Заменяя криволинейные участки адиабат, в силу их малости, отрезками прямых, найдем площадь цикла как площадь параллелограмма:
A = m(V ′−V ′)dp, |
(22.2) |
2 |
1 |
|
где V1′ и V2′ – соответственно удельные объемы жидкости и пара. Согласно теореме Карно, КПД данного цикла может быть вы-
числен как
С другой стороны, используя определение КПД цикла и выражение (22.2), можем записать, что
|
η = |
Q −Q |
= |
A |
= |
m(V2′−V1′)dp |
. |
(22.4) |
|
1 2 |
|
|
|
Q |
mλ |
|
|
Q |
|
|
|
|
|
|
1 |
|
1 |
|
|
|
|
Приравнивая правые части выражений (22.3) и (22.4), получаем
|
dp |
= |
1 λ |
. |
(22.5) |
|
|
|
|
|
|
|
dT |
T V2′−V1′ |
|
|
|
|
Это и есть уравнение Клапейрона – Клаузиуса. Оно определя-
ет угол наклона кривой фазового равновесия при фазовых переходах первого рода.
Отметим, что знак производной в уравнении (22.5), т. е. характер наклона, определяется знаком теплоты перехода и знаком разности соответствующих удельных объемов. Например, для воды линия плавления имеет отрицательный наклон, так как плотность льда меньше плотности воды и, следовательно, V2′−V1′< 0.

§ 3. Уравнение Ван-дер-Ваальса
Нидерландский физик И. Д. Ван-дер-Ваальс в 1873 г. ввел в
уравнение Клапейрона – Менделеева поправки, позволившие использовать это уравнение для описания состояния реальных газов. Эти поправки учитывают объем самих молекул газа и силы взаимодействия между ними.
Вспомним, что уравнение Клапейрона – Менделеева имеет следующий вид:
Для того, чтобы в ( 22.6) учесть «собственный» объем, занимаемый сами молекулами, нужно вначале ввести понятие эффективного диаметра молекулы.
Под эффективным диаметром молекулы реального газа понимается кратчайшее расстояние, на которое могут сблизиться две молекулы при своем взаимодействии, рассматриваемом как столкновение.
|
|
|
Из рис. 22.3 ясно, что эффективный диаметр |
|
|
|
молекулы вдвое превышает радиус действия |
|
|
|
|
|
|
сил межмолекулярного отталкивания, обуслов- |
|
|
|
ленных взаимной поляризацией молекул при |
|
|
|
их сближении друг с другом: dэфф = 2r . Тогда |
dэфф |
|
«собственный» объем, занимаемый одной мо- |
|
|
|
лекулой, |
Рис. 22.3. Объем парного |
Vi = |
1 |
3 |
столкновения молекул |
6 |
πdэфф . |
|
|
При столкновении двух молекул недоступным для других молекул является объем
V2 = 43πdэфф3 ,
из которого на одну молекулу приходится лишь половина этой величины: V1 =V2 / 2. На все молекулы в одном моле газа недоступный объем составляет, таким образом, величину
b = N V = |
4πd 3 |
N |
A |
= 4V N |
A |
. |
(22.7) |
A 1 |
6 |
эфф |
|
|
i |
|
|
|
|
|
|
|
|
|
|
|
Величина b называется постоянной |
Ван-дер-Ваальса, опреде- |
ляющей поправку к объему одного моля газа в уравнении Клапейрона – Менделеева. Для каждого реального газа она своя.
Кроме сил отталкивания, действующих на близких расстояниях, между молекулами реального газа, находящимися на расстояниях, несколько превышающих их эффективный диаметр, действуют силы притяжения. Это приводит к увеличению давления газа и необходимости введения соответствующей поправки в (22.6).
Дополнительное давление пропорционально квадрату концентрации молекул, так как оно обусловлено силами притяжения каждой из молекул, находящихся в единице объема газа, всех остальных молекул из того же объема, и поэтому должно умножаться на число этих молекул. В случае одного моля реального газа, это дополнительное давление
279
где a – постоянная Ван-дер-Ваальса, определяющая поправку на дополнительное давление к одному молю газа в уравнении Клапейрона – Менделеева (для каждого реального газа своя).
С учетом поправок a и b уравнение (22.6) принимает вид
|
2 a |
|
(V −νb)=ν RT, |
|
p +ν |
|
|
|
|
(22.9) |
V |
2 |
|
|
|
|
|
где ν – число молей газа. Уравнение (22.9) называется уравнением Ван-дер-Ваальса, а газ, который оно описывает, принято называть ван-дер-ваальсовым газом.
Ограничимся, для простоты, в дальнейшем рассмотрением одного моля ван-дер-ваальсова газа (ν =1) и перепишем уравнение
(22.9) в виде |
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
RT |
2 |
|
a |
|
ab |
|
|
V |
|
− b + |
|
V |
|
+ |
|
V − |
|
= 0. |
(22.10) |
|
p |
|
p |
p |
|
|
|
|
|
|
|
|
|
Это кубическое относительно V уравнение, имеющее в общем случае три корня. Их легко увидеть на семействе изотерм ван-дер- ваальсова газа (рис. 22. 4), когда одному и тому же давлению соответствуют три различных значения объема (точки В, D и F).
p 
А |
|
K |
Г |
|
|
|
Ж |
|
Ж - П E |
F |
|
|
|
B |
|
|
D |
П |
|
|
|
|
C |
G |
V
Рис. 22.4. Изотермы Ван-дер-Ваальса





