

Плазмохимическое осаждение, моделирование роста покрытий в условиях бомбардировки высокоскоростными атомами
Очевидно, что управление процессами эпитаксиальной и тонкопленочной технологии на качественно новом уровне позволило бы отказаться от необходимости использования макроскопических монокристаллов при получении СБИС. Ряд новых проблем возникает при решении задачи распространения метода молекулярно-лучевой эпитаксии (МЛЭ) на наноразмерный уровень, т.е. при решении проблемы создания заданных наноструктур в заданной точке подложки. В современных достаточно сложных установках для МЛЭ узконаправленные атомные пучки обычно не используются, т.е. под МЛЭ понимается динамический вариант эпитаксиальной технологии. Таким образом, на уровне лабораторного и технологического эксперимента проблема распространения методов МЛЭ и ТПТ на наноразмерный уровень к настоящему времени не решена, хотя некоторые наноструктуры, например германиевые островки (пирамиды), могут формироваться и с помощью обычных установок для МЛЭ. Однако в последнем случае речь идет о неконтролируемом (с точки зрения пространственной локализации) образовании большого количества указанных наноструктур на макроскопической подложке. Для разработки нанотехнологических аналогов методов МЛЭ и ТПТ целесообразно предварительное осуществление такой технологии на уровне компьютерного эксперимента с тем, чтобы выяснить оптимальные условия формирования наноструктур из атомного пучка. В этой области исследований имеются отдельные интересные работы. Однако они связаны либо с изучением эволюции уже построенных на твердой поверхности эпитаксиальных слоев и тонких пленок, либо с воспроизведением полудинамической ситуации, когда порция атомов, находящихся в некотором цилиндрическом сосуде, достигает его основания – поверхности твердого тела.
Одна из основных предпосылок применения методов компьютерного моделировани для изучения физических систем состоит в том, что в вычислительных экспериментах устраняются ограничения, присущие аналитическим моделям. Методы компьютерного моделирования в молекулярной физике базируются на первых принципах молекулярно-кинетической теории вещества. Они позволяют визуализировать структуру и динамику систем взаимодействующих частиц и идеально приспособлены для исследования микроскопических объектов, состоящих из 10-106 объектов и, как правило, недоступных реальному эксперименту.
Динамика классических взаимодействующих частиц описывается согласно законам движения Ньютона системой обыкновенных дифференциальных уравнений второго порядка
![]() (8.1)
(8.1)
Процедура моделирования эволюции во времени системы классических взаимодействующих частиц, определения мгновенных значений функций координат и импульсов и усреднения их по времени в соответствии с (8.1) называется методом молекулярной динамики (МД).
Динамика классических взаимодействующих частиц описывается согласно законам движения Ньютона системой обыкновенных дифференциальных уравнений второго порядка
![]() (8.2)
(8.2)
где Fi- сила, действующая на i-ю частицу в N -частичной системе. В эквивалентной форме, через два уравнения первого порядка, система записывается как
![]() (8.3)
(8.3)
![]() (8.4)
(8.4)
Вид уравнений соответствует системе, у которой энергия E – интеграл движения. Соответственно, характеристики системы вычисляются в рамках микроканонического ансамбля, для которого const NVE = const.
Потенциал межчастичного взаимодействия
Важнейшей компонентой компьютерной модели является потенциал межчастичного взаимодействия. Феноменологическое введение потенциальной функции позволяет использовать в расчетах простые аналитические выражения, содержащие несколько параметров связанных с экспериментальными данными.
Рассмотрим модельный потенциал Леннарда-Джонса (ЛД), часто используемый в вычислительном эксперименте для описания взаимодействия сферически симметричных частиц. Первоначально он был предложен для инертных газов однако часто используется для описания металлов и других типов твердых тел и жидкостей. Чаще всего он применяется в форме
 . (8.5)
. (8.5)
Параметр определяет глубину потенциальной ямы и соответствует ε энергии взаимодействия двух частиц, предоставленных самим себе. Параметр определяет положение нуля потенциала (рис. 8.1). Первый член опи- σсывает отталкивание частиц, второй - притяжение. При больших межчастичных расстояниях потенциал асимптотически стремится к нулю, при малых - к бесконечности.
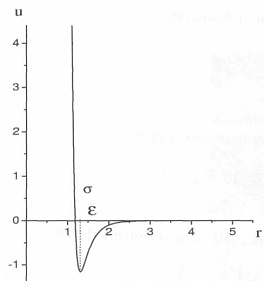
Рис. 8.1
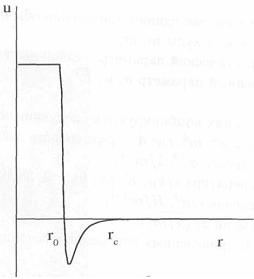
Рис. 8.2
Для использования в модели потенциал обрезается слева и справа (рис.8. 2) и приобретает вид
 (8.6)
(8.6)
Обрезание слева при r0 ≈ 0,8σ позволяет избежать очень сильного отталкивания при случайном сближении частиц на расстояние меньше r0, атакже последующей тепловой катастрофы. Слабые взаимодействия при rc>r рассчитывать нерационально, так как их можно учесть поправкой (8.9). В качестве rc обычно выбирают положение второго rc ≈ 2,8, третьего rc ≈ 3,8 и т.д. минимума радиальной функции распределения g(r) (рис. 6.5), т.е. непосредственно включаются во взаимодействие частицы двух, трех и т.д. координационных сфер.
Функция радиального распределения
![]() (8.7)
(8.7)
где
![]() (8.8)
(8.8)
число частиц в шаровом слое (r, dr).Тогда энергия взаимодействия N частиц базовой ячейки с окружением в интервале радиусов (r0, ∞) определяется как
 (8.9)
(8.9)
Задание начальных условий
При расчете методом МД всем молекулам задаются начальные положения и скорости. Начальные положения частиц могут выбираться в соответствии с планируемым фазовым состоянием модели, например в узлах кристаллической решетки. Если модельная ячейка имеет кубическую форму, число узлов в решетке должно быть равно
![]() , (8.10)
, (8.10)
где n - натуральное число.

Рис. 6.5
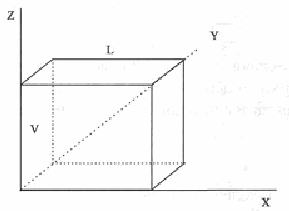
Рис. 6.6
Пусть требуется провести вычислительный эксперимент при фиксированной плотности . Поместим N частиц модельной системы в кубическую ячейку, три ребра которой совмещены с осями координат (рис.6.6). куба легко определяется из соотношений
![]() . (8.11)
. (8.11)
Численная плотность определяется выражением
![]() . (8.12)
. (8.12)
Приведенные единицы
Как правило, вычисления в моделях выполняются в приведенных единицах. Это, во-первых, позволяет избежать операций с числами разных порядков, которые вносят дополнительные погрешности; во-вторых, результаты моделирования можно легко перевести в размерный вид с использованием параметров конкретного вещества, если его межмолекулярные взаимодействия описываются использованным в модели потенциалом.
В качестве единиц приведения обычно используются:
− масса молекулы m, кг;
− энергетический параметр потенциала ε , Дж;
− линейный параметр σ, м.
Из них комбинируются следующие единицы:
− объем σ3, м3;
− плотность σ–3, м–3;
− температура /kb, К;
− давление /3, H/м2;
− время
![]() ,
с.
,
с.
В приведенных единицах потенциал ЛД записывается в виде
 . (8.13)
. (8.13)
Таким образом, метод МД основан только на первых принципах молекулярно-кинетической теории и заданном потенциале межчастичного взаимодействия. Выбор последнего в значительной степени определяет модель: класс моделируемых веществ, детальность учета взаимодействий в пределах данного класса веществ, степень количественного воспроизведения свойств прототипа модели.
Алгоритм Бимона
На основе анализа различных численных схем решения уравнений движения Бимоном (D. Beeman) была предложена схема предсказания корректировки третьего порядка, обеспечивающая наилучшие результаты с точки зрения выполнения закона сохранения энергии и затрат времени
![]() , (8.14)
, (8.14)
![]() . (8.15)
. (8.15)
Последовательность шагов алгоритма Бимона:
1.
Задать положения частиц
![]() ;
;
2.
Задать скорости частиц
![]() ;
;
3.
Задать ускорения частиц
![]() (можно положить равными нулю);
(можно положить равными нулю);
4.
Вычислить ускорения
![]() (можно положить равными нулю);
(можно положить равными нулю);
5. Вычислить новые положения частиц в момент в соответствии с (8.14);
6. Вычислить ускорения в момент ;
7. Вычислить скорости в момент в соответствии с (8.15);
8. Рассчитать мгновенные значения функций динамических переменных;
9.
Переобозначить координаты, скорости и
ускорения частиц
![]() ,
,
![]() ,
,
![]() ,
,
![]() ;
;
10. Идти к шагу 5.
Кроме перечисленных схем для численного интегрирования уравнений движения применяются схемы более высокого порядка.
Открытая система, представленная ростовой зоной в виде полусферы (показана серым цветом на рис. 1), включающей источник материальных частиц и подложку (рис. 1). Очевидно, такая модель наиболее адекватна как имеющимся, так и гипотетическим установкам для осуществления технологии МЛЭ и ТПТ.
Открытый характер системы требует более детальной проверки и оптимизации работы термостата ( метод термостата Берендсена).

Рис. 1. Схематическое изображение установки, воспроизводимой в компьютерных экспериментах.
Вместе с тем, поскольку на протяжении одного шага моделирования ростовую область покидают лишь единичные атомы, данная модель может рассматриваться как квазистационарная, что позволяет использовать обычные алгоритмы метода изотермической молекулярной динамики. С точки зрения статистической физики, ростовая область отвечает большому каноническому ансамблю Гиббса. Вместе с тем, динамический характер моделируемых процессов все же создает ряд дополнительных трудностей. В частности, оптимальная энергия (температура) пучка существенно превышает оптимальную температуру термостатирования подложки. Возникает проблема, связанная с выбором оптимального расстояния от подложки, при котором начинается ее термостатирующее действие. В данной работе эта проблема решалась с учетом локального окружения каждого из атомов, т.е. термостатирование начиналось, когда вокруг рассматриваемого атома начинала формироваться первая координационная сфера. Блок-схема алгоритма представлена на рис. 2.
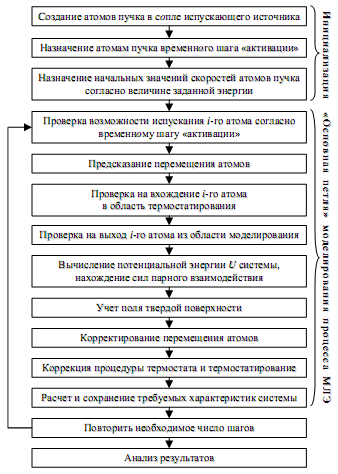
Рис. 2 Блок-схема алгоритма моделирования процесса МЛЭ.
На
начальном этапе задаются геометрические
параметры области моделирования,
выбираются диаметр источника испускания,
число атомов пучка, испущенных за один
импульс, энергия, а также частота их
испускания. Также предоставляется
возможность варьировать параметры
атомов пучка и подложки, такие как масса,
эффективный диаметр, энергетичес-кие
параметры. Далее задаются временные
характеристики и параметры термостатирования.
Каждому атому назначается шаг «активации»,
то есть порядковый номер временного
шага, на котором данный атом будет
испущен источником и «реально» появится
в системе. До данного момента атомы в
«сопле» испускающей трубы существуют
лишь «виртуально», т.е. не участвуют в
физическом взаимодействии. Шаг «активации»
назначается исходя из входного параметра
– «времена задержки между испусканиями».
Далее каждому атому задается начальная
скорость
![]() в направлении, нормальном подложке (ось
OZ):
в направлении, нормальном подложке (ось
OZ):
![]() .
.
где
![]() – случайная величина, не превышающая
по величине 25% скорости атома в заданном
направлении при энергии пучка
– случайная величина, не превышающая
по величине 25% скорости атома в заданном
направлении при энергии пучка
![]() .
.
После проведения инициализации начинается собственно продуктивная фаза моделирования, состоящая из нескольких последовательных действий. Взаимно противоречащие требования к точности решения системы уравнений второго закона Ньютона и практические соображения приемлемого времени счета приводят к компромиссному значению временного шага δt порядка 10–14 с = 0,01 пс. Элементарный шаг («основная петля») эволюции осуществляется с помощью «модифицированного алгоритма скоростей Верле»:
![]() ,
,
![]() ,
,
![]() ,
,
где
t
– текущий момент времени,
,
![]() ,
,
![]() – радиус-вектор i-го
атома, скорость и ускорение соответственно.
– радиус-вектор i-го
атома, скорость и ускорение соответственно.
При моделировании в рамках модели парных взаимодействий был использован потенциал Леннард-Джонса:
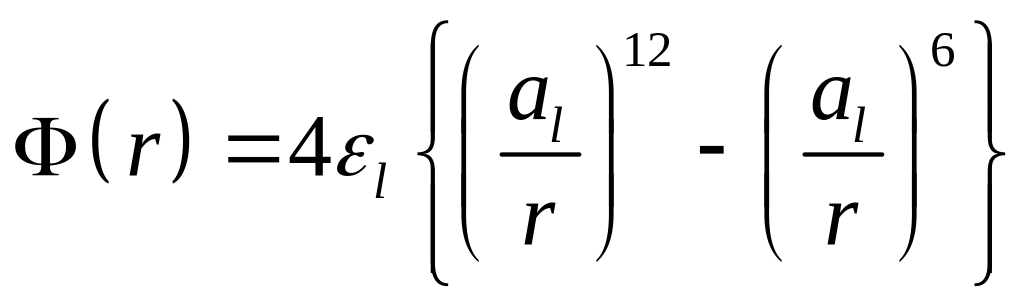 ,
,
где r – расстояние между атомами эффективного диаметра al, εl – энергетический параметр (глубина потенциальной ямы).
Еще одну достаточно сложную проблему представляет корректный учет ван-дер-ваальсовых сил, действующих на наночастицу со стороны твердого тела. Первое приближение к потенциалу подложки можно получить исходя из допущения о том, что твердое полупространство ограничивается континуальной потенциальной стенкой. В случае леннард-джонсовской подложки для потенциальной энергии атома жидкости, находящегося на расстоянии z от континуальной твердой поверхности, получим:
 ,
,
где
as
и εs
– соответствующие параметры парного
потенциала, описывающего взаимодействие
в твердом полупространстве,
![]() – приведенная плотность числа атомов
в твердом полупространстве.
– приведенная плотность числа атомов
в твердом полупространстве.
Ниже приводятся результаты, связанные с изучением влияния параметров исследуемой системы на морфологические и структурные характеристики формирующихся нанокристаллов.
Энергия пучка. Основным параметром атомного пучка является его энергия, т.е. средняя кинетическая энергия атомов. В реальных системах, в зависимости от энергии пучка материальных частиц, возможно протекание различных по физической природе процессов – от формирования нанокристаллов (энергии от 0,5 до 10 эВ) до ионной имплантации (энергии порядка 1 КэВ и выше) и ядерных реакций (энергии, превышающие 10 ГэВ). Если принять, что в существующих испарителях, применяющихся в МЛЭ и ТПТ температура испаряемого вещества не превышает (2000-5000) K , то данному интервалу будут соответствовать максимально достижимые энергии (0,3-0,7) эВ. Однако как видно из рис. 3, при энергии пучка порядка 0,5 эВ имеет место гомогенная конденсация, т.е. образование кластеров в самом пучке еще до достижения атомами подложки. По-видимому, при использовании метода вакуум-термического испарения на макроскопическом уровне, атомы располагаются в пространстве с меньшей плотностью, и такого рода эффект гомогенной конденсации проявляется в меньшей степени. Кроме того, гомогенное образование нанокластеров не является существенным препятствием для получения микроструктур и тонких пленок. Однако для решения проблемы получения наноструктур из атомного пучка в заданной точке подложки эффект гомогенной конденсации следует исключить, что можно достичь увеличением энергии пучка. Вместе с тем, при энергиях пучка порядка 0,5 КэВ (рис. 4), т.е. при высоких энергиях, проявляется другой «паразитный» эффект – существенно возрастает распыление формирующихся наноструктур (см. рис. 4 и рис. 5а). В результате проведенных компьютерных экспериментов было установлено, что оптимальным условиям формирования наноструктур отвечают энергии пучка порядка 5 эВ (рис. 5б).

Рис. 3 Формирование зародыша твердой фазы в пучке.

Рис.4 Распыление структуры, формирующейся на твердой подложке.
Как
видно из рис. 6, при оптимальной энергии
пучка (3-5 эВ), на поверхности континуальной
подложки наблюдается формирование
слоистых наноструктур (плоскости слоев
параллельны твердой поверхности). Как
и следовало ожидать, гораздо большая
упорядоченность и пирамидоподобная
морфология наблюдаются при низких
температурах (рис. 6а). Приведенная
температура
![]() определяется через глубину потенциальной
ямы парного потенциала
определяется через глубину потенциальной
ямы парного потенциала
![]() парного потенциала εl. Как показали
предварительные компьютерные эксперименты
и теоретические оценки, температуре
плавления леннард-джонсовских наночастиц
отвечает T*=0,5. Для конфигураций, показанных
на рис. 6, относительный энергетический
параметр подложки
парного потенциала εl. Как показали
предварительные компьютерные эксперименты
и теоретические оценки, температуре
плавления леннард-джонсовских наночастиц
отвечает T*=0,5. Для конфигураций, показанных
на рис. 6, относительный энергетический
параметр подложки
![]() равен единице, что соответствует случаю,
который в теории эпитаксиального роста
принято называть автоэпитаксией.
равен единице, что соответствует случаю,
который в теории эпитаксиального роста
принято называть автоэпитаксией.
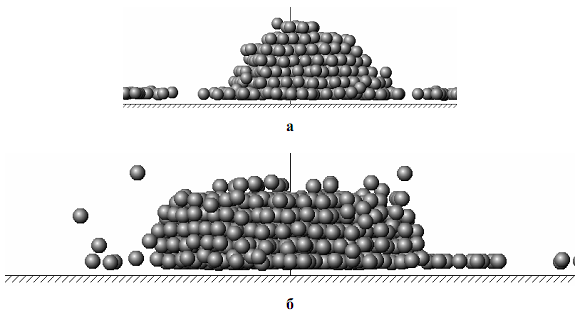
Рис. 6. Конечные конфигурации наноструктур N=1000 и энергии пучка 5 эВ:
а) T* = 0,2; б) T* = 0,4
После активной фазы нанесения атомов пучка на подложку протекает процесс релаксации сформированной наноструктуры. Кинетические зависимости для первого координационного числа z1 для систем, отвечающих рис. 6, представлены на рис. 7.
Для случая, когда T*=0,2 (рис. 7а), наблюдаются незначительные флуктуации z1 в диапазоне 11,9÷12,0 на протяжении времени релаксации, отвечающего горизонтальной асимптоте. Приведенное время t* отвечает числу элементарных шагов моделирования. Одному элементарному шагу соответствует время порядка 10–14 с (0,01 пс). Точное соответствие с реальным временем зависит от массы атома и энергетического параметра потенциального взаимодействия атомов, из которых состоит пучок.
Принимая во внимание выраженную огранку, слоистую пирамидоподобную форму и высокое значение z1 можно сделать вывод о том, что конфигурацию, представленную на рис. 6а, можно рассматривать как малодефектный пирамидоподобный нанокристалл. При T*=0,4 (рис. 6б) огранка не проявляется, хотя слоистость сохраняется, а морфология наночастицы отвечает куполообразной форме. На стадии релаксации флуктуации z1 становятся более заметными (в диапазоне от 10 до 12) и не уменьшаются при увеличении времени релаксации. Это позволяет предположить, что случаю, представленному на рис. 6б отвечает аморфная наноструктура.
Влияние
дискретности подложки.
Рис. 9 соответствует наноструктурам,
сформировавшимся на дискретных подложках
с ГПУ-структурой, являющихся аналогами
континуальной поверхности с относительным
энергетическим параметром
![]() =
1 (этому случаю отвечают конфигурации,
показанные на рис. 6). Сравнение
конфигураций, показанных на рис. 6 и рис.
9, кинетических зависимостей для
коэффициента распыления и радиальных
функций (рис. 8 и рис. 10) не выявляет
существенных различий между конденсацией
на континуальной и дискретной подложках,
что представляется на первый взгляд
довольно неожиданным. Предложенное
объяснение сводится к тому, что на
континуальной подложке формируется
нижний монослой атомов (прекурсор),
который задает структуру вышележащих
слоев. Иными словами, основная роль
подложки связана не с ее атомной
структурой, а с тем, что плоская поверхность
задает ориентацию кристаллографических
граней, параллельных плоскости подложки.
Данный вывод согласуется с результатами
лабораторных экспериментов Дж. Бернала,
в которых изучалась структура объектов,
полученных «насыпкой» металлических
шаров на плоскую твердую поверхность
(Бернал Дж. // Рост кристаллов. Т. 5. М.:
Наука, 1965. С. 149).
=
1 (этому случаю отвечают конфигурации,
показанные на рис. 6). Сравнение
конфигураций, показанных на рис. 6 и рис.
9, кинетических зависимостей для
коэффициента распыления и радиальных
функций (рис. 8 и рис. 10) не выявляет
существенных различий между конденсацией
на континуальной и дискретной подложках,
что представляется на первый взгляд
довольно неожиданным. Предложенное
объяснение сводится к тому, что на
континуальной подложке формируется
нижний монослой атомов (прекурсор),
который задает структуру вышележащих
слоев. Иными словами, основная роль
подложки связана не с ее атомной
структурой, а с тем, что плоская поверхность
задает ориентацию кристаллографических
граней, параллельных плоскости подложки.
Данный вывод согласуется с результатами
лабораторных экспериментов Дж. Бернала,
в которых изучалась структура объектов,
полученных «насыпкой» металлических
шаров на плоскую твердую поверхность
(Бернал Дж. // Рост кристаллов. Т. 5. М.:
Наука, 1965. С. 149).
Влияние температуры подложки. На рис. 11 приведены конечные конфигурации, полученные после испускания источником N=1000 атомов и отвечающие достаточно широкому температурному интервалу T*=0,2÷0,5.
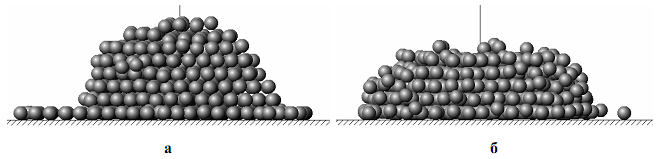
Рис. 9. Наноструктуры, сформированные на подложке с гексагональной плотноупакованной структурой (N=1000, энергия пучка 5 эВ, энергетический параметр подложки равен 1): а) Т*=0,2; б) Т*=0,4.

Рис. 11. Наноструктуры, полученные на континуальной подложке, N=1000, энергия пучка 5 эВ, энергетический параметра атомов подложки равен 1: а) Т*=0,2; б) Т*=0,3; в) Т*=0,4; г) Т*=0,5.
Как следует из рис. 11, при низких температурах (T*=0,2) на подложке формируются слоистые пирамидоподобные нанокристаллы, обладающие специфической огранкой, наблюдающейся также у германиевых островков (Пчеляков О.П. и др. // Физика и техника полупроводников. 2000. Т. 34. Вып. 11. С. 1281). При дальнейшем повышении температуры до T*=0,3 и T*=0,4, происходит снижение степени упорядоченности и наблюдается переход от к куполообразной форме.
Влияние
энергии подложки.
Еще одним управляющим фактором, влияющим
на морфологию и структуру наночастиц,
формирующихся из атомного пучка, является
энергетический параметр подложки
![]() .
Под
понимается
энергетический параметр парного
потенциала, т.е. глубина потенциальной
ямы. В первом приближении, этот параметр
пропорционален энергии связи для
конденсированной фазы, отвечающей
данному химическому элементу.
.
Под
понимается
энергетический параметр парного
потенциала, т.е. глубина потенциальной
ямы. В первом приближении, этот параметр
пропорционален энергии связи для
конденсированной фазы, отвечающей
данному химическому элементу.
Влияние диаметра атомного пучка и количества атомов, испущенных за один импульс. Меняя диаметр пучка, можно обеспечивать формирование структур определенной формы и радиуса в заданной точке подложки. Выбор оптимального значения диаметра пучка должен быть тесно связан с оптимизацией числа атомов N, испущенных за один импульс. Чем меньше N, тем меньший диаметр пучка d необходимо задавать для получения квазиравновесных пирамидоподобных нанокристаллов. При неадекватно больших значениях диаметра атомного пучка и малом числе испущенных атомов, наблюдается формирование лишь 1-2 упорядоченных (эпитаксиальных) слоев на поверхности континуальной подложки.
Влияние выбора потенциала межатомного взаимодействия. Представленные выше результаты получены с использованием парного потенциала Леннард-Джонса, одного из наиболее простых и удобных парных потенциалов, позволяющего адекватно учесть соотношение между силами межатомного отталкивания и притяжения. Очевидно, результаты, связанные с влиянием отмеченных выше управляющих параметров на морфологию и структуру формирующихся нанокристаллов, должны иметь достаточно общий характер. Вместе с тем, парные потенциалы типа потенциала Леннард-Джонса отвечают достаточно сильному межатомному притяжению, результатом которого является формирование плотноупакованных структур. Наиболее адекватно потенциал Леннард-Джонса описывает благородные газы в сжиженном и твердом состоянии, в несколько худшем приближении – металлы, формирующие плотную упаковку. В соответствии с полученными нами результатами, леннард-джонсовские нанокристаллы характеризуются ГПУ-структурой, которая приводит к формированию гексагонального основания леннард-джонсовских пирамид. К сожалению, переход от парных потенциалов к многочастичным увеличивает время счета в десятки раз и существенно ограничивает величину числа атомов, испускаемых за один импульс.