

ГЛАВА 57. ВРОЖДЁННЫЕ ПОРОКИ РАЗВИТИЯ
57.1. НЕОНАТАЛЬНЫЙ СКРИНИНГ НА НАСЛЕДСТВЕННУЮ И ВРОЖДЁННУЮ ПАТОЛОГИЮ
Врождённые пороки развития (ВПР) считают важнейшей медицинской и социальной проблемой, поскольку они занимают ведущее место в структуре причин перинатальной, неонатальной и младенческой заболеваемости, смертности и инвалидности. Согласно ВОЗ, ВПР отмечают у 4–6% детей. В России ежегодно более 50 000 детей рождаются с ВПР, число пациентов с ВПР превышает 1,5 млн человек. Высокие затраты на лечение, уход и реабилитацию детей с ВПР обусловливают необходимость разработки и совершенствования методов контроля, диагностики и профилактики ВПР у детей.
КОД ПО МКБ-10
Q00–Q99 Врождённые аномалии (пороки развития), деформации и хромосомные нарушения.
ЭПИДЕМИОЛОГИЯ
Частота рождения детей с ВПР зависит от внешних средовых и мультифакториальных факторов, развития общества и его возможностей профилактики и пренатальной диагностики ВПР. Очень важно проводить эти мероприятия среди всего населения репродуктивного возраста.
КЛАССИФИКАЦИЯ
В зависимости от действия вредных факторов в период беременности ВПР подразделяют на следующие типы. ·Гаметопатии или мутации в половых клетках родителей, в том числе возрастные, которые проявляются наследственными синдромами и заболеваниями.
·Бластопатии возникают в первые 15 сут после оплодотворения яйцеклетки и проявляются двойниковыми пороками, сиреномиелиями.
·Эмбриопатии представлены в виде всех пороков, которые возникают в период от 16 сут до 8 нед беременности. ·Фетопатии развиваются в период развития плода от 9 нед до конца беременности в виде дистопий и гипоплазии органов.
Выделяют изолированные (при локализации в одном органе), системные и множественные ВПР, малые аномалии развития, которые не приводят к изменению жизненных функций ребёнка и не ограничивают его деятельность.
В международных системах генетического мониторинга проводят регистрацию ВПР по нозологической форме и коду согласно МКБ-10.
ЭТИОЛОГИЯ
Возникновение пороков развития более чем в 60% случаев связано с мультифакториальными причинами, в 20% — неустановленными, в 5–6% случаев — другими причинами (моногенными, хромосомными и внешнесредовыми). Под термином «ВПР» обозначают структурные дефекты, а под термином «врождённая аномалия» — структурные, функциональные и метаболические нарушения органа или участка тела.
ДИАГНОСТИКА
Во многих странах мира существуют национальные регистры ВПР, предназначенные для контроля и профилактики ВПР. Их объединяет международная неправительственная организация ICBDMS, созданная в 1974 г. для обмена информацией, определения частоты ВПР в мире и проведения научных исследований.
Ведение национального регистра помогает проводить своевременную коррекцию ВПР у детей в раннем возрасте и способствует предупреждению развития инвалидности.
ИНСТРУМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
УЗИ считают наиболее информативным методом диагностики ВПР плода. Его эффективность зависит от уровня профессиональной подготовки специалистов и разрешающей возможности современных аппаратов.
СКРИНИНГ
Неонатальный скрининг предполагает массовое обследование новорождённых на врождённые и наследственные заболевания. В мировой практике проводят скрининг 8–12 заболеваний. В России с 1994 г. в родильных стационарах путём пункции пятки новорождённого осуществляют забор крови для диагностики врождённого гипотиреоза и фенилкетонурии. В 2006 г. скрининговая программа была дополнена исследованиями для выявления ВГКН, галактоземии и муковисцидоза.
Заболевания, выявляемые при неонатальном скрининге, протекают тяжело и при отсутствии своевременной диагностики и адекватного лечения приводят к глубоким и необратимым изменениям, нередко с летальным исходом, развитием умственной недостаточности и тяжёлой патологии. При этих заболеваниях ранняя диагностика и коррекция выявленных нарушений на протяжении жизни позволяют избежать неблагоприятных последствий.
Проведение скрининга не должно препятствовать ранней выписке из стационара.
Служба неонатального скрининга — многоуровневая организационная структура. Родовспомогательные учреждения у всех новорождённых проводят забор крови, которой пропитывают участок бумажного бланка. Их отправляют в медикогенетические лаборатории для определения маркеров заболевания. В случае положительного результата одной из проб, показаны дополнительные исследования для подтверждения или исключения заболевания.
Важно определить место пункции кожи на пятке новорождённого для забора крови при проведении неонатального скрининга. Место пункции располагается медиальнее линии, проведённой от I пальца до пятки или латеральнее от линии, проведённой от границы между IV и V пальцами до пятки (рис. 57-1). Необходимо согреть предполагаемое место пункции тёплой водой (с температурой до 41 °С) в течение 3–5 мин, протереть место предполагаемой пункции тампоном, смоченным этиловым спиртом. Затем следует хорошо просушить это место стерильной марлей, во избежание гемолиза.
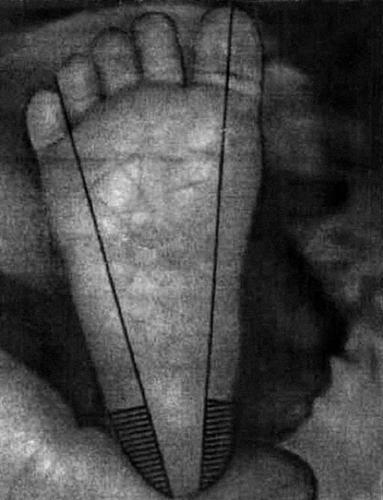
Рис. 57-1. Определение места пункции на коже пятки новорождённого.
ЛЕЧЕНИЕ
Специализированная медицинская педиатрическая служба (генетическая, эндокринологическая и др.) занимается лечением больных детей. Эффективность функционирования этой многоэтапной системы определяется быстрой диагностикой и началом лечения ребенка в доклинический период и зависит от слаженности взаимодействия всех её участников.
ПРОФИЛАКТИКА
Роль медицинской генетики в формировании здоровья будущих поколений считают основополагающей. Одним из важных её достижений служит профилактика ВПР плода путём массового назначения фолиевой кислоты в период, предшествующий зачатию и в первые месяцы беременности. Данный метод профилактики приводит к снижению риска рождения детей с пороками головного и спинного мозга.
Пренатальную диагностику ВПР плода считают вторичной профилактикой. С целью выявления тяжёлых и летальных ВПР с неблагоприятным прогнозом для жизни ребенка, во время беременности проводят УЗИ (3 раза), забор материала для исследований (амниоцентез, кордоцентез, биопсия хориона) и цитогенетические и биохимические исследования.
ОПИСАНИЕ ВРОЖДЁННЫХ ПОРОКОВ РАЗВИТИЯ
АНОМАЛИИ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ АНЭНЦЕФАЛИЯ
Один из наиболее частых пороков развития ЦНС, при котором отсутствуют полушария мозга и свод черепа. При экзэнцефалии отсутствуют также кости свода черепа, но имеется фрагмент мозговой ткани. Акрания характеризуется отсутствием свода черепа, при наличии аномально сформированного головного мозга.
КОД ПО МКБ-10
Q00 Анэнцефалия.
ЭПИДЕМИОЛОГИЯ
Частота возникновения данного порока различна в разных регионах мира.
ЭТИОЛОГИЯ
Анэнцефалия — результат нарушения закрытия рострального отдела нейропоры в течение 28 дней с момента оплодотворения. С помощью динамических ультразвуковых исследований установлено, что акрания, экзэнцефалия и анэнцефалия — этапы развития одного порока.
Анэнцефалия и акрания — пороки мультифакториальной природы. Анэнцефалия может входить в состав синдрома амниотических тяжей, сочетаться с хромосомными аберрациями (трисомия 18, кольцевая хромосома 13), возникать в результате действия химиотерапии, на фоне диабета и гипертермии матери.
Описанные пороки абсолютно летальны.
ДИАГНОСТИКА
Пренатальная диагностика возможна уже в I триместре беременности.
ЦЕФАЛОЦЕЛЕ
Цефалоцеле представляет собой выход мозговых оболочек через дефект костей черепа. В случаях, когда в состав грыжевого мешка входит мозговая ткань, аномалия носит название энцефалоцеле. Если в состав входит часть желудочковой системы, используют термин «менингоэнцефалоцистоцеле». Наиболее часто дефекты располагаются в области затылка, но могут возникать и в других отделах (лобном, теменном, назофарингеальном).
КОД ПО МКБ-10
Q01 Цефалоцеле.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости аномалии составляет 1 случай на 2000 живорождённых. При наличии цефалоцеле нередко встречаются другие аномалии развития головного мозга:
●при больших размерах грыжевого мешка создаются условия для развития микроцефалии;
●часто диагностируют вентрикуломегалию и гидроцефалию;
●в 7–15% случаев выявляют spina bifida.
Энцефалоцеле часто входит в состав многочисленных синдромов: Уокера–Варбурга, криптофтальма, диссегментарной дисплазии, Нормана–Робертса, Гольденхара и др.
ЭТИОЛОГИЯ
Цефалоцеле развивается в результате незаращения дефекта нервной трубки и возникает на 4 нед внутриутробной жизни.
ДИАГНОСТИКА
Пренатальная диагностика энцефалоцеле во II и III триместрах беременности, как правило, не вызывает затруднений.
ТАКТИКА ВРАЧА
При выявлении цефалоцеле следует предложить прерывание беременности по медицинским показаниям. При продлении беременности тактика родоразрешения зависит от размеров и содержимого грыжевого мешка. Кесарево сечение рекомендовано при наличии маленького дефекта и маленьких размеров грыжевого мешка.
ВРОЖДЁННАЯ ГИДРОЦЕФАЛИЯ
Это собирательный термин, обозначающий избыточное расширение ликворосодержащих пространств головного мозга. Для этого состояния характерно увеличение размеров желудочков мозга с одновременным нарастанием внутричерепного давления, сопровождающимся (в большинстве случаев) увеличением размеров головы.
КОД ПО МКБ-10
Q03 Врождённая гидроцефалия.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости гидроцефалии составляет от 0,1 до 2,5 случаев на 1000 новорождённых, при этом изолированные формы диагностируют с частотой 0,39–0,87 на 1000 новорождённых, в 60% случаев аномалия возникает у мальчиков.
В подавляющем большинстве случаев (до 99%) формируется внутренняя гидроцефалия, для которой характерно расширение желудочков. Внутренняя гидроцефалия чаще обструктивная, то есть обусловленная наличием препятствия на пути оттока ликвора. Причина 43% всех гидроцефалий — стеноз водопровода мозга.
ЭТИОЛОГИЯ
Гидроцефалия — следствие первичных пороков отдельных структур мозга, но может возникнуть вторично под влиянием многочисленных анте-, интра- и постнатальных факторов (инфекции, тератогены, опухоли, травмы, гипоксия).
КЛАССИФИКАЦИЯ
По топическому признаку различают три основных варианта аномалии:
●внутренняя гидроцефалия — увеличение желудочковой системы плода;
●наружная гидроцефалия — увеличение субарахноидальных пространств;
●смешанная гидроцефалия — одновременное расширение желудочков и субарахноидальных пространств мозга. При вовлечении в процесс обоих полушарий мозга гидроцефалию называют двусторонней.
ДИАГНОСТИКА
Высокочувствительным эхографическим симптомом служит увеличение высоты тела желудочка на 2–3 мм по сравнению с нормальными значениями этого показателя.
Пренатальное обследование должно включать кариотипирование, вирусологические тесты, тщательное ультразвуковое исследование с подробной оценкой структур спинного мозга и позвоночника, опорно-двигательной системы и кистей плода. Родителям необходима консультация нейрохирурга для определения возможностей лечения в неонатальном периоде. При выявлении гидроцефалии до периода жизнеспособности плода целесообразно обсудить вопрос о прерывании беременности.
СИНДРОМ ДЕНДИ–УОКЕРА
Для этой патологии характерно нарушение развития ромбовидного мозга, включающее частичную или полную агенезию червя мозжечка, кистозное расширение IV желудочка мозга и формирование кисты в области субарахноидального пространства задней черепной ямки.
КОД ПО МКБ-10
Q03.1 Синдром Денди–Уокера.
ЭПИДЕМИОЛОГИЯ
Среди живорождённых детей частота встречаемости синдрома Денди–Уокера относительно невысокая — 1 случай на 25 000–35 000. Среди детей, страдающих гидроцефалией, синдром Денди–Уокера диагностируют значительно чаще — в 3,5–12,0% случаев.
ДИАГНОСТИКА
Синдром Денди–Уокера — одно из немногих патологических состояний плода, при котором результаты пренатального кариотипирования служат абсолютным показанием. Хромосомную патологию при данном синдроме выявляют в 29– 55% случаев, изменения генома представлены, в подавляющем большинстве случаев, грубыми аберрациями.
ТАКТИКА ВРАЧА
Прогноз для жизни и здоровья при синдроме Денди–Уокера зависит от наличия сочетанных аномалий развития, хромосомных аберраций и срока диагностики. В тех случаях, когда диагноз синдрома Денди–Уокера удаётся поставить до периода жизнеспособности плода, пациентке следует предложить прерывание беременности.
МИКРОЦЕФАЛИЯ
Это клинический синдром, для которого типично уменьшение массы и размеров головного мозга, сопровождающееся неврологическими расстройствами и нарушения умственного развития.
КОД ПО МКБ-10
Q02 Микроцефалия.
ЭПИДЕМИОЛОГИЯ
Встречается с частотой 1,6 на 1000 новорождённых.
ТАКТИКА ВРАЧА
Микроцефалия — неизлечимое заболевание, поэтому при установлении диагноза до достижения плодом жизнеспособного возраста следует предложить прерывание беременности, однако, пренатальная диагностика данной аномалии по ультразвуковой картине представляет определённые сложности.
АНЕВРИЗМА БОЛЬШОЙ МОЗГОВОЙ ВЕНЫ
Аневризма большой мозговой вены (Галена) — сложный порок, варьирующий в размерах от гигантской аневризмы до множественных коммуникаций между системой вены Галена и мозговыми сосудами системы сонных и вертебробазилярных артерий.
ЭПИДЕМИОЛОГИЯ И ЭТИОЛОГИЯ
Частота развития данного порока неизвестна, мнения об эмбриогенезе противоречивы.
У 50% новорождённых с аневризмой вены Галена развиваются симптомы сердечной недостаточности.
ДИАГНОСТИКА
Пренатальная диагностика аневризмы вены Галена имеет важное практическое значение. Новые возможности в идентификации сосудистых мальформаций головного мозга открывает трёхмерная эхография, которая позволяет детально оценить архитектонику сосудов головного мозга.
ТАКТИКА ВРАЧА
Прогноз при этом неблагоприятный — более чем в 90% случаев наступает смерть в неонатальном периоде или младенческом возрасте.
АГЕНЕЗИЯ МОЗОЛИСТОГО ТЕЛА
Мозолистое тело представляет собой широкую срединную структуру, соединяющую полушария мозга, формирующую нижнюю стенку межполушарной борозды и крышу III желудочка мозга. Агенезия может быть полной или частичной, при которой полость III желудочка мозга остаётся открытой.
КОД ПО МКБ-10
Q04.0 Агенезия мозолистого тела.
ЭПИДЕМИОЛОГИЯ
Истинная частота агенезии мозолистого тела не известна, поскольку в некоторых случаях эта аномалия развития не имеет клинических симптомов. Наиболее частые причины агенезии — воспаление и нарушение кровообращения.
ЭТИОЛОГИЯ
Агенезия мозолистого тела — гетерогенная патология — проявление более чем 40 хромосомных аберраций, более 120 моногенных заболеваний и синдромов множественных пороков развития.
ДИАГНОСТИКА
Пренатальное обследование должно включать кариотипирование и тщательное ультразвуковое исследование, при котором особое внимание следует уделять анатомии головного мозга.
ГОЛОПРОЗЭНЦЕФАЛИЯ
Редкая летальная аномалия развития нервной трубки плода. Представляет собой сложный порок развития, для которого характерно нарушение процесса деления переднего мозгового пузыря, что приводит к отсутствию разделения конечного мозга на сферы.
КОД ПО МКБ-10
Q04.2 Голопрозэнцефалия.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости голопрозэнцефалии колеблется от 0,6 до 1,9 случаев на 10 000 живорождённых. Так как многие случаи порока спонтанно прерываются в ранние сроки гестации, предполагают, что частота возникновения данной патологии более высока — до 1 на 250 беременностей.
КЛАССИФИКАЦИЯ
В зависимости от степени и стадии нарушения процесса деления переднего мозгового пузыря W. De Mayer выделил три формы голопрозэнцефалии:
●алобарную (бездолевую);
●семилоборную (полудолевую);
●лобарную (долевую).
Пороки прозэнцефалической группы, к которым относят алобарную голопрозэнцефалию, очень часто сочетаются с аномалиями лица (гипотелоризмом, аринией, расщелинами верхней губы и нёба, циклопией, цебоцефалией, этмоцефалией, микрофтальмией). Среди экстракраниальных сочетанных аномалий отмечают кистозные дисплазии почек, костно-суставные пороки, омфалоцеле, сердечно-сосудистые аномалии, миеломенингоцеле.
ЭТИОЛОГИЯ
Большинство случаев — спорадические, этиология неизвестна. Голопрозэнцефалия обычно сочетается с хромосомными отклонениями (трисомии 13, 15, 18, 21, моносомии 13q-, 18q-, 18p-, триплоидия).
ДИАГНОСТИКА
Современные технологи позволяют не только диагностировать голопрозэнцефалию в ранние сроки гестации, но и пренатально дифференцировать различные морфотипы порока.
ТАКТИКА ВРАЧА
Прерывание беременности следует предлагать при обнаружении любых форм голопрозэнцефалии.
ИНИЭНЦЕФАЛИЯ
Редкий летальный порок развития нервной трубки плода, для которого характерны значительное укорочение шейного отдела позвоночника, незавершённое формирование основания черепа, особенно области большого затылочного отверстия, расположение части мозга в области задней черепной ямки.
КОД ПО МКБ-10
Q00.2 Иниэнцефалия.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости данной аномалии в популяции колеблется от 0,01% до 0,16%.
ГИДРОАНЭНЦЕФАЛИЯ
Это состояние, при котором большая часть полушарий мозга отсутствует и заменена спинномозговой жидкостью. Степень поражения полушарий мозга различна — часть коры в височной и затылочной областях может быть сохранена или происходит её полная деструкция.
Синонимы Гидроцефалическая анэнцефалия, гидроэнцефалодисплазия, цистэнцефалия.
КОД ПО МКБ-10
Q03.8 Гидроанэнцефалия.
ТАКТИКА ВРАЧА
Во всех случаях обнаружения гидроанэнцефалии необходимо рекомендовать прерывание беременности.
АНОМАЛИЯ АРНОЛЬДА–КИАРИ
Порок развития нервной трубки, возникающий на ранних этапах эмбриогенеза, проявляющийся смещением одной или обеих миндалин мозжечка и ствола головного мозга в большое затылочное отверстие и ниже.
КОД ПО МКБ-10
Q07.0 Синдром Арнольда–Киари.
КЛАССИФИКАЦИЯ
Патоморфологически выделяют три основные типа этой аномалии:
I — проникновение миндалин мозжечка в шейный отдел позвоночного канала;
II — вклинение дисплазированного мозжечка в большое затылочное отверстие в сочетании с удлинением ствола мозга;
III — изолированное тотальное смещение структур заднего мозга в расширенное затылочное отверстие, сопровождающееся образованием грыжи в затылочной области.
II и III типы порока характеризуются высокой летальностью в перинатальном периоде.
SPINA BIFIDA
Аномалия развития позвоночного столба, возникающая в результате нарушения процесса закрытия нервной трубки. Термин spina bifida объединяет широкий спектр дефектов — от клинически незначимого отсутствия отдельного фрагмента позвонка до незаращения целых отделов позвоночника. Закрытые дефекты составляют 15% всех дорсальных spina bifida. Обычно при этих дефектах нет других ассоциированных аномалий ЦНС.
Spina bifida часто сочетается с типичной внутричерепной патологией, включая вентрикуломегалию, гипоплазию структур задней черепной ямки.
КОД ПО МКБ-10
Q05 Spina bifida.
ЭПИДЕМИОЛОГИЯ
Spina bifida — один из наиболее распространённых пороков развития ЦНС, встречается в среднем с частотой 1– 2 случая на 1000 живорождённых. Кроме того, Spina bifida — проявление более чем 40 хромосомных аномалий и синдромов множественных пороков развития.
КЛАССИФИКАЦИЯ
●Spina bifida occulta — закрытый волокнистой тканью дефект пластинок дуг одного или большего количества позвонков. Практически не имеет клинически выраженных симптомов и выявляется случайно при инструментальном исследовании позвоночника.
●Occulta spinal dysraphism — характеризуется сращением спинного мозга в крестцовом отделе (tethered cord
syndrome), клинически проявляется нарушением сенсорной и моторной функций нижних конечностей, недержанием мочи и кала, болью, сколиозом.
ДИАГНОСТИКА
Так как головка плода легко доступна ультразвуковому исследованию, нарушения мозговой архитектоники служат вспомогательным признаком при диагностике spina bifida.
Наиболее значимые для пренатальной диагностики формы spina bifida (менингомиелоцеле, рахисхизис, менингоцеле) — пороки с яркой ультразвуковой картиной дефекта, которые можно диагностировать с 12–14 нед беременности. Пренатальная диагностика закрытых спинальных дефектов крайне затруднена в связи с сохранением эхографической картины всех структур спинного мозга, позвоночника и мягких тканей.
ТАКТИКА ВРАЧА
Spina bifida относят к дефектам, требующим прерывания беременности при диагностике до периода жизнеспособности плода в связи с неблагоприятным перинатальным прогнозом. В III триместре беременности супруги должны быть поставлены в известность о прогнозе для будущего ребёнка.
Наиболее важные вопросы относительно тактики акушерской помощи касаются срока и метода родоразрешения — оптимальным считают родоразрешение доношенного ребёнка, показанием для досрочного родоразрешения может служить быстрое нарастание сочетанной вентрикуломегалии и макрокрании.
АРАХНОИДАЛЬНЫЕ КИСТЫ
Это экстрапаренхиматозные внутричерепные тонкостенные образования, исходящие из слоёв арахноидальной оболочки.
ЭПИДЕМИОЛОГИЯ
Арахноидальные кисты — редкие аномалии развития головного мозга, на них приходится не более 1% от всех внутричерепных образований.
КЛАССИФИКАЦИЯ
Различают врождённые (первичные) и приобретённые (вторичные) арахноидальные кисты.
●Первичные кисты — результат нарушения формирования паутинной оболочки или субарахноидального пространства в ранние сроки беременности.
●Вторичные кисты развиваются в результате воспалительного процесса в мозговых оболочках, травмы или
субарахноидального кровоизлияния и диагностируются в поздние сроки беременности или после рождения.
ТАКТИКА ВРАЧА
Прогноз для жизни и здоровья при арахноидальных кистах небольших размеров, отсутствии сочетанных аномалий развития и нормальном кариотипе может быть благоприятным.
РЕДКИЕ ПОРОКИ РАЗВИТИЯ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ
К редко встречающимся формам пороков развития ЦНС относят дисгенезии (дисплазии) мозга, обусловленные аномалиями миграции нейронов — лиссэнцефалию (синдром Миллера-Дикера), пахигирию, порэнцефалию, шизэнцефалию, гидроанэнцефалию.
ПОРОКИ РАЗВИТИЯ ДЫХАТЕЛЬНОЙ СИСТЕМЫ ДИАФРАГМАЛЬНАЯ ГРЫЖА
Редкий врождённый порок, проявляющийся перемещением органов брюшной полости в грудную через дефект диафрагмы.
КОД ПО МКБ-10
Q79.0 Диафрагмальная грыжа.
ЭПИДЕМИОЛОГИЯ
Частота этого порока составляет 1 случай на 2000–5000 живорождённых.
КЛАССИФИКАЦИЯ
К основным видам врождённых диафрагмальных грыж относятся:
●заднебоковую (грыжа Бохдалека);
●парастернальную (грыжа Морганьи);
●релаксацию диафрагмы.
Грыжевой мешок чаще содержит тонкий кишечник и желудок, реже — толстую кишку и селезёнку. В некоторых случаях в грыжевое содержимое могут быть вовлечены поджелудочная железа, печень, надпочечники и даже почки. При перемещении печени в грудную полость всегда отмечается декстрокардия.
ПАТОГЕНЕЗ
Диафрагмальная грыжа служит одной из причин гипоплазии лёгких, нередко приводящей к летальному исходу. Большинство исследователей связывают неблагоприятный исход с частым наличием сочетанных аномалий, среди которых преобладают дефекты нервной трубки, расщелины лица, омфалоцеле, врождённые пороки сердца и хромосомные аберрации.
ДИАГНОСТИКА
Пренатальная эхографическая диагностика диафрагмальной грыжи возможна со второй половины беременности. По данным R. Snijders и К. Nicolaides, в среднем у 18% плодов с пренатально диагностированной диафрагмальной грыжей обнаруживают хромосомные аберрации (чаще трисомия 18).
ТАКТИКА ВРАЧА
Прогноз при врождённой диафрагмальной грыже во многом зависит от степени выраженности гипоплазии лёгочной ткани. В целом для детей с диафрагмальной грыжей, выявленной пренатально с помощью эхографии, он неблагоприятный: выживаемость новорождённых низка, а смертность достигает 73–86%.
ГИДРОТОРАКС
Это патологическое скопление жидкости, чаще лимфы, в плевральной полости.
ЭПИДЕМИОЛОГИЯ
Частота возникновения плевральных выпотов составляет в среднем 1 случай на 10 000 новорождённых. Не редко гидроторакс сочетается с врождёнными пороками развития лёгких и сердца.
ЭТИОЛОГИЯ
Патологическое скопление жидкости в плевральной полости плода — результат её гиперпродукции или низкой интенсивности реабсорбции. Снижение реабсорбции может быть связано с нарушением оттока лимфы от лёгких или с патологией лимфатических сосудов.
Умеренный гидроторакс чаше — односторонний процесс. При выраженном гидротораксе лёгкие плода резко гипоплазированы, оттеснены к стенкам грудной клетки или органам средостения.
ТАКТИКА ВРАЧА
Прогноз при пренатально выявленном гидротораксе неблагоприятный. Большинство исследователей сообщают о практически 100% перинатальной смертности.
ВРОЖДЁННЫЙ КИСТОЗНО-АДЕНОМАТОЗНЫЙ ПОРОК РАЗВИТИЯ ЛЁГКИХ ЭТИОЛОГИЯ
Кистозно-аденоматозный порок — гамартома лёгких, характеризующуюся разрастанием конечных отделов бронхиол с формированием мешотчатых образований. Гамартома лёгких относится к доброкачественным опухолевидным образованиям. Формирование этой аномалии происходит до 5 нед гестации. Кистозно-аденоматозному пороку развития лёгких часто сопутствует многоводие, плацентомегалия и неиммунная водянка плода.
ТАКТИКА ВРАЧА
Акушерская тактика зависит от наличия сочетанных пороков развития, неиммунной водянки, смещения органов средостения и многоводия, так как эти факторы прогностически неблагоприятные.
ЛЁГОЧНАЯ СЕКВЕСТРАЦИЯ
Врожденный порок развития, при котором часть аномально развитой легочной паренхимы отделена от неизменённого легкого. Секвестрированная часть не связана с воздухоносными путями и получает кровь посредством артерий, отходящих от аорты. При этом отсутствует и функциональный кровоток в секвестрированном лёгком.
Синонимы Добавочное лёгкое, бронхопульмональная секвестрация.
КОД ПО МКБ-10
Q33.2 Лёгочная секвестрация.
КЛАССИФИКАЦИЯ
●При расположении участка с аномальным кровоснабжением внутри нормально развитого лёгкого говорят об интралобарной секвестрации, которая чаще локализуется в нижней доле правого лёгкого.
●Добавочное лёгкое с аортальным кровоснабжением, расположенное вне нормально развитого лёгкого (в грудной полости, на шее, в брюшной полости и др.), называют экстралобарной лёгочной секвестрацией.
●Кроме этого, возможно наличие интралобарной и экстралобарной экстраторакальной локализации секвестра.
ТАКТИКА ВРАЧА
Лёгочная секвестрация нередко сочетается с другими аномалиями, прежде всего с бронхопульмональными пороками, диафрагмальной грыжей, врождёнными пороками сердца, неиммунной водянкой плода. В этих случаях прогноз для плода крайне неблагоприятный.
БРОНХОГЕННЫЕ КИСТЫ КЛАССИФИКАЦИЯ
●При нарушении на ранних этапах формирования трахеи и бронхов развиваются кисты, располагающиеся в области трахеи, пищевода, бифуркации трахеи или главных бронхов, то есть в пределах средостения.
●При более поздних нарушениях развития кисты могут располагаться внутри лёгких.
Чаще возникают одиночные бронхогенные кисты.
ЭТИОЛОГИЯ
Бронхогенные кисты возникают в результате нарушения развития трахеобронхиального дерева в эмбриональном периоде. Их локализация и гистологическая структура зависит от времени нарушения развития трахеобронхиального дерева.
ДИАГНОСТИКА
Пренатальная диагностика бронхогенных кист вызывает значительные трудности, так как многие внутригрудные аномалии имеют схожую эхографическую картину. Значительную помощь в верификации диагноза бронхогенной кисты оказывает сканирование в режиме ЦДК и трёхмерной реконструкции.
АНОМАЛИИ ПЕРЕДНЕЙ БРЮШНОЙ СТЕНКИ ГАСТРОШИЗИС
Гастрошизис — эвентрация органов брюшной полости (чаще кишечника) через параумбиликальный дефект передней брюшной стенки, располагающийся обычно справа от пупка. Грыжевые органы при этом не имеют мембраны.
КОД ПО МКБ-10
Q79.3 Гастрошизис.
ЭПИДЕМИОЛОГИЯ
Гастрошизис встречается с частотой 0,94 на 10 000 новорождённых.
КЛАССИФИКАЦИЯ
Выделяют изолированную и сочетанную формы порока.
●Изолированный гастрошизис возникает чаше (79%), преимущественно у молодых матерей.
●Сочетанную форму выявляют в 10–30% случаев. Она представляет собой комбинацию гастрошизиса с атрезией или стенозом кишечника.
ЭТИОЛОГИЯ
Гастрошизис возникает в результате преждевременной инволюции правой пупочной вены или разрыва дистального сегмента омфаломезентериальной артерии, что приводит к ишемии тканей правой околопупочной области и развитию дефекта.
ДИАГНОСТИКА
Точность ультразвуковой диагностики гастрошизиса во II и III триместрах беременности варьирует от 70 до 95%. Большинство случаев изолированного гастрошизиса спорадические. Риск повторного развития данной аномалии низкий. Частота выявления хромосомных аномалий при данной патологии не превышает популяционную, в связи с этим от пренатального кариотипирования можно воздержаться.
ТАКТИКА ВРАЧА
При наличии у плода гастрошизиса в III триместре беременности необходимо проводить динамическую оценку его функционального состояния, поскольку вероятность развития дистресс-синдрома достаточно высока, а в 23–50% случаев формируется задержка внутриутробного развития плода.
Прогноз для новорождённых с изолированным гастрошизисом благоприятный: выживают более 90% детей. В связи с этим его пренатальное обнаружение не служит показанием для прерывания беременности. Родоразрешение путём операции кесарева сечения не оказывает существенного влияния на постнатальную заболеваемость и смертность, а родоразрешение через естественные родовые пути не сопровождается ухудшением течения постнатального периода.
ОМФАЛОЦЕЛЕ
Это дефект передней брюшной стенки в области пупочного кольца с образованием грыжевого мешка с внутрибрюшинным содержимым, покрытого амниоперитонеальной мембраной.
ЭПИДЕМИОЛОГИЯ
Патология встречается с частотой 1 случай на 3000–6000 новорождённых.
ЭТИОЛОГИЯ
Порок возникает в результате невозвращения органов брюшной полости через пупочное кольцо. Дефект локализуется по средней линии живота и может быть центральным, эпи- и гипогастральным. Чаще всего грыжевым содержимым оказываются петли кишечника, желудок и печень, покрытые мембраной, состоящей из двух слоёв: внутреннего (брюшина) и наружного (амнион). Размеры дефекта могут быть от очень небольших до 12 см.
Сочетанные пороки развития при омфалоцеле встречаются в 61,1% случаев, хромосомные аномалии выявляют в 7,5– 54,0% случаев. Хромосомные дефекты включают трисомии 13, 18, 21 и моносомию 45Х0.
ДИАГНОСТИКА
Диагностика омфалоцеле не представляет каких-либо сложностей, она возможна уже в I триместре беременности.
ТАКТИКА ВРАЧА
Мнения относительно метода родоразрешения различны. Большинство авторов считают, что показанием для родоразрешения путём кесарева сечения служат большие размеры омфалоцеле.
АНОМАЛИЯ РАЗВИТИЯ СТЕБЛЯ ТЕЛА
Абсолютно летальный порок, относящийся к дефектам передней брюшной стенки, обусловленным неправильным развитием краниальной, каудальной и латеральной закладок туловища эмбриона в ранние сроки гестации. Этот порок характеризуется обширным дефектом передней брюшной стенки, выраженным кифосколиозом и рудиментарной пуповиной, верхняя половина туловища плода располагается в амниотической полости, нижняя — в целомической.
ЭПИДЕМИОЛОГИЯ
Частота развития аномалии составляет в среднем 1 случай на 14 000 новорождённых.
ДИАГНОСТИКА
Значительные анатомические изменения плода при аномалии развития стебля тела позволяют диагностировать её уже в конце I триместра беременности.
ПОРОКИ РАЗВИТИЯ МОЧЕВЫДЕЛИТЕЛЬНОЙ СИСТЕМЫ ДВУХСТОРОННЯЯ АГЕНЕЗИЯ ПОЧЕК
Отсутствие почек — летальная аномалия мочевыделительной системы. Синонимы Синдром Поттер 0.
КОД ПО МКБ-10
Q60.1 Двусторонняя агенезия почек
ЭПИДЕМИОЛОГИЯ
Частота встречаемости двухсторонней агенезии почек составляет 1–3 случая на 10 000 новорождённых с преимущественным поражением плодов мужского пола.
ДИАГНОСТИКА
Синдром Поттер 0 можно диагностировать при эхографии в начале II триместра беременности. Диагностические критерии — отсутствие ультразвуковой тени почек и мочевого пузыря, а также выраженное маловодие, обычно сопровождающееся симметричной задержкой развития плода.
Односторонняя почечная агенезия встречается значительно чаще, однако диагностируется только в единичных случаях. Это объясняется тем, что при односторонней почечной агенезии, как правило, сохраняется нормальное
количество околоплодных вод, визуализируется мочевой пузырь, надпочечник может быть принят за почку. |
|
|
ТАКТИКА ВРАЧА |
|
|
Синдром Поттер 0 служит абсолютным показанием для прерывания беременности. |
|
|
ПОЛИКИСТОЗНАЯ БОЛЕЗНЬ ПОЧЕК ИНФАНТИЛЬНОГО ТИПА |
|
|
Синонимы |
|
|
Синдром Поттер I. |
|
|
КОД ПО МКБ-10 |
|
|
Q61.1 Поликистозная болезнь почек инфантильного типа |
|
|
ЭПИДЕМИОЛОГИЯ |
|
|
Инфантильный тип поликистоза почек плода, |
по данным E.L. Potter, встречается редко — 2 случая |
на |
110 000 новорождённых. |
|
|
ЭТИОЛОГИЯ |
|
|
Поликистозная болезнь почек инфантильного |
типа — летальный двухсторонний порок, развивающийся |
в |
эмбриональном периоде при несвоевременном и неправильном соединении секреторной и экскреторной частей почек. При этом не происходит полноценное развитие почечных канальцев и отсутствует их связь с собирательными трубками. Одна часть почечных канальцев запустевает, другая превращается в ретенционные необструктивные кисты. Чашечно-лоханочная система почки при этом остаётся интактной. Почки плода при этом пороке поражаются симметрично, кистозные повреждения представлены «полостными» образованиями размерами 1–2 мм. Заболевание наследуется по аутосомно-рецессивному типу.
ДИАГНОСТИКА
К эхографическим критериям поликистозной болезни почек инфантильного типа относят:
●двустороннюю нефромегалию;
●отсутствие ультразвуковой тени мочевого пузыря;
●выраженное маловодие.
Эхогенность почек при данном пороке значительно превышает эхогенность окружающих тканей («гиперэхогенные почки», «большие белые почки»).
Диагностика синдрома Поттер I возможна с середины II триместра беременности.
МУЛЬТИКИСТОЗНАЯ ДИСПЛАЗИЯ ПОЧЕК
Синонимы Синдром Поттер II.
ЭПИДЕМИОЛОГИЯ
Наиболее частая форма почечной дисплазии.
ДИАГНОСТИКА
Процесс чаще всего носит односторонний характер. При синдроме Поттер II поражённая почка значительно увеличена в размерах, привычная форма и нормальная ткань её отсутствуют, паренхима представлена множественными кистами с анэхогенным содержимым. Размеры кист варьируют в широком диапазоне.
Ультразвуковая диагностика мультикистозной дисплазии почек возможна со второй половины беременности.
ТАКТИКА ВРАЧА
При двустороннем процессе показано прерывание беременности, при одностороннем — возможно продление гестации.
В случаях одностороннего выраженного увеличения почки, приводящего к значительному увеличению размеров живота плода и компрессии органов брюшной полости, возможна внутриутробная пункция почечных кист под эхографическим контролем или установка шунта.
ПОЛИКИСТОЗНАЯ БОЛЕЗНЬ ПОЧЕК ВЗРОСЛОГО ТИПА
Синонимы Синдром Поттер III.
КОД ПО МКБ-10
Q61.2 Поликистозная болезнь почек взрослого типа.
ЭПИДЕМИОЛОГИЯ
Поликистоз почек взрослого типа встречается значительно чаще синдрома Поттер 0, однако, его пренатальная диагностика, весьма затруднена, в большинстве случаев заболевание проявляется у взрослых.
ЭТИОЛОГИЯ
Аутосомно-доминантное заболевание, проявляющееся замещением паренхимы почки множественными кистами разного диаметра вследствие расширения собирательных канальцев нефрона.
ДИАГНОСТИКА
Процесс чаще носит односторонний характер, мочевой пузырь всегда визуализируется, количество околоплодных вод соответствует норме или несколько уменьшено.
ТАКТИКА ВРАЧА
При обнаружении поликистоза взрослого типа в I и II триместрах целесообразно прерывание беременности, тогда как его диагностика в III триместре беременности не требует изменения стандартной акушерской тактики.
КИСТОЗНАЯ ДИСПЛАЗИЯ ПОЧЕК
Синонимы Синдром Поттер IV.
КОД ПО МКБ-10
Q61.4 Кистозная дисплазия почек.
ЭТИОЛОГИЯ
Заболевание возникает вторично по отношению к обструкции мочевыводящих путей, наблюдаемой в ранние сроки. Наиболее частые причины, вызывающие выраженное изменение почек, — задние уретральные клапаны и атрезия уретры.
ДИАГНОСТИКА
Почки могут быть увеличены в размерах с множественными мелкими, обычно периферическими, кистами. При синдроме Поттер IV, как и при остальных клинических формах синдромов Поттер, часто возникает выраженное маловодие.
ОБСТРУКТИВНЫЕ УРОПАТИИ КОД ПО МКБ-10
Q62 Обструктивные уропатии.
ЭПИДЕМИОЛОГИЯ
Данная патология занимает лидирующее место среди всех аномалий мочевыделительной системы плода. Особо выделяют пиелоэктазии, для которых характерно транзиторное течение. По данным Н.П. Веропотвеляна и соавт., частота пиелоэктазий, выявленных пренатально, составляет 21,7 на 1000 плодов, при этом в 45,6% диагностируют двустороннюю пиелоэктазию.
Гидронефроз пренатально выявляют значительно реже — 2,8 случая на 1000 плодов. Контроль за состоянием почек с середины II триместра почти в 70% случаев позволяет выявить спонтанную редукцию пиелоэктазии к концу беременности или к окончанию периода новорождённости.
ЭТИОЛОГИЯ
Широко распространено мнение, что пиелоэктазия — начальный признак гидронефротической трансформации почек. При изолированной пиелоэктазии частота хромосомных аномалий составляет в среднем 3%, а при наличии сочетанных аномалий — 24%. Риск обнаружения хромосомных дефектов приблизительно одинаков как при одностороннем, так и при двустороннем гидронефрозе. Однако частота хромосомных дефектов почти в 2 раза выше у плодов женского пола (18%) в сравнении с плодами мужского пола (10%).
ДИАГНОСТИКА
По мнению большинства исследователей, динамическому наблюдению, детальной оценке анатомии и пренатальному кариотипированию подлежат все плоды, у которых во II триместре беременности переднезадний размер почечной лоханки при поперечном сканировании превышает 4 мм, в III триместре — 7 мм.
ТАКТИКА ВРАЧА
С целью декомпрессии ткани паренхимы почки показано шунтирование или пункция лоханки плода. При последующей коррекции в постнатальном периоде эта операция позволяет добиться сохранения функции почки.
МЕГАУРЕТЕР
Мегауретер — расширение мочеточника, которое может сопровождаться дилатацией почечной лоханки.
КОД ПО МКБ-10
Q62.2 Мегауретер.
КЛАССИФИКАЦИЯ
Выделяют три основные формы расширения мочеточника — мегауретер, связанный с рефлюксом, обусловленный обструкцией, а так же мегауретер без рефлюкса и обструкции.
●Мегауретер, вызванный рефлюксом, чаще обусловлен аномалиями пузырно-мочеточникового соустья.
●Мегауретер, связанный с обструкцией, — чаще результат аномалий дистальных отделов мочевых путей.
●Мегауретер без обструкции и рефлюкса представляет собой идиопатическое расширение мочеточника над
пузырным соустьем.
ЭТИОЛОГИЯ
В большинстве случаев мегауретер — вторичное состояние по отношению к патологии дистальных отделов мочевых путей.
ОБСТРУКТИВНЫЕ УРОПАТИИ НА НИЗКОМ УРОВНЕ
Чаще вызывают аномалиями уретры, такие как атрезия, агенезия, стриктуры, стеноз и клапаны.
ЭТИОЛОГИЯ
Наиболее частая причина — задние уретральные клапаны, на долю которых приходится 38% всех наблюдений.
ДИАГНОСТИКА
Эхографическая картина характеризуется наличием резко расширенного мочевого пузыря, мегауретера, гидронефроза и маловодия у плодов, как правило, мужского пола.
СИНДРОМ PRUNE BELLY
Синонимы
Синдром триады, синдром Eagle-Barrett.
ЭПИДЕМИОЛОГИЯ
Редкий порок развития — встречается с частотой 1 случай на 35 000–50 000 новорождённых, преимущественно страдают мальчики.
ДИАГНОСТИКА
Для синдрома Prune Belly характерны три основных признака:
●перерастяжение передней брюшной стенки;
●резко увеличенный в размерах гипотоничный мочевой пузырь;
●крипторхизм.
При обнаружении синдрома Prune Belly оправдано пренатальное кариотипирование, так как проявления синдрома могут наблюдаться в комплексе врождённых пороков развития плода при хромосомных аномалиях (чаще при трисомии 18 или 21 хромосомы), когда другие эхографические маркеры могут остаться не распознанными из-за резко выраженного маловодия.
ТАКТИКА ВРАЧА
В случае определения нормального кариотипа плода тактика ведения беременности зависит от выраженности и длительности маловодия, степени вовлечения в патологический процесс мочевыделительной и других систем плода.
ЭКСТРОФИЯ МОЧЕВОГО ПУЗЫРЯ КОД ПО МКБ-10
Q64.1 Экстрофия мочевого пузыря.
ЭПИДЕМИОЛОГИЯ
Частота возникновения экстрофии мочевого пузыря составляет в среднем 1 случай на 30 000–50 000 новорождённых, соотношение мальчиков и девочек 2,3:1,0.
ЭТИОЛОГИЯ
Это редкий порок развития, связанный с неправильным развитием каудальной закладки передней брюшной стенки. При этой аномалии отсутствует передняя стенка мочевого пузыря, а задняя расположена снаружи
ДИАГНОСТИКА
Основные критерии диагноза:
●мочевой пузырь не выявляется при УЗИ при неизменённых почках и нормальном количестве околоплодных вод;
●низкое прикрепление пуповины;
●низкая брюшная выпуклость, заключающая в себе экстрофированный мочевой пузырь;
●аномалии гениталий.
ТАКТИКА ВРАЧА
Сочетание поражения мочевого пузыря, уретры, костей таза, позвоночника, половых органов определяет сложность лечения, его многоэтапность и трудоёмкость, а порой и невозможность обеспечения удовлетворительного качества жизни детей с этой аномалией, поэтому при пренатальном обнаружении целесообразно прерывание беременности.
АНОМАЛИИ РАЗВИТИЯ ЛИЦА И ШЕЙ МИКРОФТАЛЬМИЯ И АНОФТАЛЬМИЯ
Микрофтальмия — уменьшение размера глазных яблок, анофтальмия — их отсутствие. Однако этот диагноз следует считать прерогативой патологоанатомов, которые должны обнаружить не только отсутствие глазных яблок, но и зрительных нервов, зрительного перекрёста и зрительных путей. Микро- и анофтальмия — причина 4% случаев врождённой слепоты.
ДИАГНОСТИКА
Микрофтальмия может быть заподозрена при диаметре глазницы менее 5 процентиля для соответствующего гестационного возраста.
При подозрении на микрофтальмию показано комплексное тщательное обследование в центре пренатальной диагностики для исключения сочетанных аномалий и хромосомных дефектов, так как микрофтальмия часто входит в состав множественных врождённых пороков развития и различных синдромов.
ЦИКЛОПИЯ
Эта аномалия редко бывает изолированной, чаще всего она сочетается с другими врождёнными пороками развития или входит в состав различных синдромов.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости циклопии в среднем составляет 1 случай на 40 000 живорождённых.
ДИАГНОСТИКА
Пренатальный диагноз циклопии устанавливают на основании обнаружения у плода единственного глаза или частично разделённых глазных яблок в одной (общей), чаще всего центрально расположенной, орбите.
ДАКРИОЦИСТОЦЕЛЕ
КЛАССИФИКАЦИЯ
Дакриоцистоцеле может быть как односторонним, так и двухсторонним.
ЭТИОЛОГИЯ
Дакриоцистоцеле представляет собой кисту слёзного протока, которая возникает в результате нарушения строения складки Гаснера и функции ямки Розенмюллера.
ДИАГНОСТИКА
Пренатальная ультразвуковая диагностика кисты слёзного протока основывается на обнаружении гипоэхогенных образований округлой формы, локализующихся ниже и медиальнее орбит. Обычно диаметр дакриоцистоцеле не превышает 10–12 мм, а структура представлена однородным содержимым.
ТАКТИКА ВРАЧА
Кисты слёзного протока не приводят к изменению орбит и часто спонтанно исчезают как во внутриутробном периоде, так и после рождения. Поэтому в случаях обнаружения дакриоцистоцеле следует придерживаться выжидательной тактики, ограничиваясь только динамическим наблюдением.
ВРОЖДЁННАЯ КАТАРАКТА ЭПИДЕМИОЛОГИЯ
Встречается с частотой 1,2–9,4 случая на 10 000 новорождённых.
ЭТИОЛОГИЯ
Эта патология глазного яблока может возникать в результате недостаточной функции паращитовидной железы у матери или плода. Причиной также могут быть перенесённые во время беременности матерью инфекционные заболевания, чаще TORCH-комплекс, разнообразные врождённые ферментативные нарушения. Врождённая катаракта — компонент многих наследственных синдромов.
ДИАГНОСТИКА
Пренатальная диагностика врождённой катаракты основана на обнаружении у плода повышенной эхогенности хрусталика, что возможно уже во II триместре беременности. Следует помнить, что визуализация нормального хрусталика не позволяет исключить врождённую катаракту.
РАСЩЕЛИНЫ ЛИЦА
Расщелины лица — аномалии развития, которые, будучи изолированными, не влияют на прогноз для жизни и здоровья ребёнка и хорошо поддаются хирургической коррекции. С другой стороны они входят в состав многочисленных наследственных синдромов, нередко служат фенотипическим проявлением хромосомных аномалий, сочетаются с пороками развития практически всех органов и систем.
КОД ПО МКБ-10
Q35–37 Расщелины лица.
КЛАССИФИКАЦИЯ
В зависимости от степени тяжести и локализации порока различают:
● одноили двухсторонние расщелины верхней губы (полные, частичные), сопровождающиеся деформацией носа и асимметрией его крыльев; ● расщелины верхней губы и нёба, распространяющиеся на альвеолярный отросток, твёрдое и мягкое нёбо.
● срединные и боковые расщелины (одно или двухсторонние).
В большинстве наблюдений расщелина губы сочетается с расщелиной нёба, однако в клинической практике регистрируют также случаи изолированной локализации аномалии.
ПРОБОСЦИС
Пробосцис — хоботообразный отросток с одним или двумя внутренними отверстиями.
КОД ПО МКБ-10
Q30 Пробосцис.
ЭПИДЕМИОЛОГИЯ
Довольно редкая аномалия — 1 случай на 16 000–40 000 новорождённых.
КЛАССИФИКАЦИЯ
Хоботообразный отросток может располагаться в нижней части лба (этмоцефалия) или в области обычного расположения носа (цебоцефалия) и, как правило, имеет одно центральное отверстие.
АРИНИЯ
Ариния — отсутствие носа.
КОД ПО МКБ-10
Q30 Ариния.
ЭТИОЛОГИЯ
Причина развития аринии в большинстве случаев неизвестна. Может встречаться изолированно или как часть комплекса пороков.
ТАКТИКА ВРАЧА
Прогноз и акушерская тактика зависят от наличия и формы сочетанных аномалий. Изолированная ариния совместима с жизнью и не требует изменения стандартной акушерской тактики.
МАКРОГЛОССИЯ
Это выраженное увеличение языка, которое обычно сопровождает синдром Бекуита–Видеманна, болезнь Дауна, гипотиреоз.
ДИАГНОСТИКА
Заключение о наличие макроглоссии может быть сделано на основании постоянного нахождения языка за пределами ротовой полости.
ОТОЦЕФАЛИЯ
Очень редкий летальный врождённый порок развития, этиология и частота которого неизвестны. Этот порок развития характеризуется уродливым изменением черт лица, что обусловлено отсутствием или гипоплазией нижней челюсти, сближением височных костей и горизонтальным расположением ушных раковин.
Синонимы Синотия, мелотия.
КИСТОЗНАЯ ГИГРОМА ШЕИ
Кистозная гигрома — аномалия лимфатической системы, проявляющаяся одиночными или множественными кистозными образованиями мягких тканей шейной области.
КЛАССИФИКАЦИЯ
Кистозная гигрома чаще всего бывает многокамерной, двусторонней и локализуется кзади и сбоку от шеи.
Следует отметить, что локализацию гигромы в шейном отделе отмечают в 75% случаев, кроме того, это образование может «распространяться» в нижележащие отделы туловища, подмышечную впадину (20%), полость рта, средостение (3–10%). В 2% наблюдений гигрома может располагаться в брюшной полости или ретроперитонеально, в 2% случаев — достигать паховой области и нижних конечностей.
При кистозной гигроме часто обнаруживают хромосомные аномалии и некоторые генетические синдромы.
ДИАГНОСТИКА
Обязательно проводят пренатальное кариотипирование.
ПОРОКИ СЕРДЦА И МАГИСТРАЛЬНЫХ СОСУДОВ
Врождённые пороки сердца — одни из самых распространённых аномалий развития, они встречаются с частотой 7– 12 случаев на 1000 новорождённых. Врождённые пороки сердца привлекают пристальное внимание исследователей во всём мире не только в связи с высокой встречаемостью, но и потому, что служат основной причиной смерти детей на первом году жизни от пороков развития. Среди детей, рождающихся с врождёнными пороками сердца, 14–29% умирают в первую неделю жизни, 19–42% — в течение первого месяца, а 40–87% младенцев не доживают до конца первого года.
АНОМАЛИИ РАСПОЛОЖЕНИЯ СЕРДЦА КОД ПО МКБ-10
Q24.0 Декстрокардия.
КЛАССИФИКАЦИЯ
Сердце новорождённого может неправильно располагаться в средостении и полости перикарда, встречается так же и эктопия сердца.
К первой группе аномалий положения сердца относят декстрокардию и мезокардию.
●При декстрокардии сердце располагается большей частью справа от средней линии тела. Верхушка сердца находится справа от грудины.
●Мезокардия — положение сердца, при котором его ось лежит в средне-сагиттальной плоскости.
Эктопия сердца — редкий врождённый порок развития, при котором сердце частично или полностью располагается за пределами грудной клетки.
Различают четыре основных типа эктопии: грудная, торакоабдоминальная, абдоминальная и шейная.
●При грудной эктопии сердце располагается вне грудной полости, выходя наружу через дефект грудины. При этой форме эктопии возникает дефект грудины, отсутствует париетальная часть перикарда, а грудная клетка имеет маленькие размеры.
●Торакоабдоминальную эктопию чаще рассматривают как вариант синдрома Кантрелла, она включает пять
сочетанных аномалий:
◊дистальный дефект грудины;
◊срединный дефект брюшной стенки в надпупочной области;
◊диафрагмальную грыжу;
◊недостаточность диафрагмальной части перикарда;
◊врождённые интракардиальные дефекты, чаще межжелудочковой перегородки. ● Шейные эктопии характеризуются смещением сердца в шейную область.
● При абдоминальном типе эктопии сердце смещено в брюшную полость через первичный дефект диафрагмы.
ЭТИОЛОГИЯ
Неправильное положение сердца в средостении в пренатальном периоде чаще всего связано с врождённой диафрагмальной грыжей, плевральным выпотом и кистозным аденоматозным пороком развития левого лёгкого.
ДЕФЕКТЫ ПЕРЕГОРОДОК СЕРДЦА КОД ПО МКБ-10
Q21 Дефекты интракардиальных перегородок.
ЭПИДЕМИОЛОГИЯ
Данные аномалии — одни из наиболее частых разновидностей врождённых пороков сердца (20–30%).
Клинически значимые |
дефекты межпредсердной перегородки |
возникают |
с частотой 0,24–0,58 на |
1000 новорождённых, а |
дефекты межжелудочковой перегородки — |
0,38–2,26 на |
1000 новорождённых. Частота |
развития мелких гемодинамически незначимых дефектов межжелудочковой перегородки значительно выше и составляет свыше 50 случаев на 1000 новорождённых, большинство из них спонтанно закрывается к 1–2 году жизни. Частота возникновения общего предсердно-желудочкового канала сердца составляет 0,15–0,44 на 1000 новорождённых.
Гипопластический синдром левых отделов сердца составляет 7–9% от всех врождённых пороков сердца у живорождённых детей и оказывается одной из наиболее частых причин смерти.
Частота возникновения единственного желудочка — в среднем 1,5% от всех врождённых пороков сердца.
КЛАССИФИКАЦИЯ Открытое овальное окно — один из наиболее частых пороков, выявляемых в постнатальном периоде.
Диагностировать его в пренатальном периоде невозможно, так как во внутриутробной жизни оно служит физиологическим шунтом.
Общий предсердно-желудочковый канал представляет собой группу сердечных аномалий, включающую дефекты межжелудочковой и межпредсердной перегородок с расщеплением атриовентрикулярных клапанов. До 50% случаев общего предсердно-желудочкового канала сочетается с анэуплоидией (трисомия 21 — 60%, трисомия 18 — 25%). Различают две формы порока: полную и неполную.
●При неполной форме возникает разделение атриовентрикулярных клапанов, обычно существует связь между предсердиями или между левым желудочком и правым предсердием. Правый атриовентрикулярный клапан чаще сформирован правильно, левый обычно имеет три створки, а между передней и задней створками имеется щель.
●Полная форма характеризуется сочетанием первичных дефектов межпредсердной и межжелудочковой перегородок с расщеплением створок обоих атриовентрикулярных клапанов. При этом дефекты перегородок сливаются и образуют
общий атриовентрикулярный клапан, ограниченный фиброзным кольцом, несущим пять створок.
Гипопластический синдром левых отделов сердца — группа аномалий, характеризующихся недоразвитием левого желудочка с атрезией или выраженной гипоплазией митрального и/или аортального клапанов. Нередко при этом пороке также выявляют атрезию или гипоплазию аорты, при этом основная лёгочная артерия может быть значительно расширена.
Единственный желудочек сердца — тяжёлый врождённый порок, при котором желудочки сердца представлены единой камерой.
ДИАГНОСТИКА
Пренатальная эхокардиографическая диагностика полной формы острого предсердно-желудочкового канала или единственного желудочка обычно не вызывает трудностей.
ТАКТИКА ВРАЧА
Прогноз при остром предсердно-желудочковом канале в большинстве случаев неблагоприятный, так как этот порок часто сочетается с другими аномалиями и хромосомными дефектами. В этих случаях оправдано прерывание беременности.
АТРЕЗИЯ ТРЁХСТВОРЧАТОГО КЛАПАНА
Атрезия трёхстворчатого клапана сердца характеризуется отсутствием тока крови через правый атриовентрикулярный канал и маленькими размерами правого желудочка, обычно с дефектом межжелудочковой перегородки. Расположение магистральных сосудов может быть различным. Нередко при атрезии трёхстворчатого клапана и гипоплазии правого желудочка отмечают атрезию или гипоплазию лёгочной артерии.
КОД ПО МКБ-10
Q22.4 Атрезия трикуспидального клапана.
ДИАГНОСТИКА
Точность пренатальной диагностики во многом зависит от качества оценки четырёхкамерного среза сердца плода при скрининговом ультразвуковом исследовании.
ТАКТИКА ВРАЧА
Прогноз при АТК неблагоприятный.
АНОМАЛИЯ ЭБШТЕЙНА Сравнительно редкий врождённый порок развития правых отделов сердца. КОД ПО МКБ-10
Q22.5 Аномалия Эбштейна. Эпидемиология
Частота возникновения аномалии Эбштейна среди всех врождённых пороков сердца варьирует в пределах 0,05–1%, и составляет в среднем 1 случай на 20 000 живорождённых.
ЭТИОЛОГИЯ
В основе аномалии Эбштейна лежит нарушение формирования септального и заднего парусов трёхстворчатого клапана, которые в этом случае развиваются непосредственно из эндокарда правого желудочка сердца. Это приводит к смещению аномального клапана вглубь правого желудочка, обусловливая его разделение на два отдела: дистальный (подклапанный) — активный, и проксимальный (надклапанный или атриализованный) — пассивный. Надклапанный отдел, соединяясь с правым предсердием, формирует единое функциональное образование. Самостоятельно этот порок сердца встречается редко и обычно сочетается с открытым овальным окном, дефектом межжелудочковой перегородки, атрезией или стенозом лёгочного ствола, тетрадой Фалло, коарктацией аорты, наджелудочковой тахикардией и др.
ТАКТИКА ВРАЧА
Очевидно, что наличие грубых сочетанных пороков и хромосомных дефектов делают прогноз при аномалии Эбштейна неблагоприятным.
ФИБРОЭЛАСТОЗ ЭНДОКАРДА
Это диффузное утолщение эндокарда, обусловленное пролиферацией фиброзной и эластической ткани, ведущее к снижению пластичности и ухудшению диастолической функции сердца. Фиброэластоз эндокарда — редкий врождённый порок сердца, в большинстве случаев приводящий к гибели в перинатальном периоде в связи со сниженной сократительной способностью миокарда.
КЛАССИФИКАЦИЯ
Поражение может быть изолированным или сочетаться с другими аномалиями.
ДИАГНОСТИКА
Пренатальная ультразвуковая диагностика возможна с начала II триместра беременности, однако фиброэластоз эндокарда далеко не во всех случаях проявляется до 20 нед беременности.
АНОМАЛИЯ УЛЯ
Редкая врождённая патология, проявляющаяся выраженной гипоплазией или почти полным отсутствием миокарда одного из желудочков сердца (чаще правого).
Синонимы Гипоплазия правого желудочка.
КОД ПО МКБ-10
Q24.8 Аномалия Уля.
КЛАССИФИКАЦИЯ
Как правило, гипоплазия бывает диффузной, хотя возможна очаговая форма. Трёхстворчатый клапан сердца может быть нормальным или дисплазированным. В некоторых случаях отмечают возникает стеноз или атрезия клапана лёгочного ствола.
ДИАГНОСТИКА
В ходе пренатального ультразвукового исследования обращает на себя внимание выраженная кардиомегалия за счёт дилатации поражённого желудочка. Нередко регистрируют признаки застойной сердечной недостаточности (перикардиальный и плевральный выпот, асцит).
ВРОЖДЁННЫЕ КАРДИОМИОПАТИИ
Под врожденными кардиомиопатиями подразумевают группу различных поражений сердечной мышцы, которые могут быть вызваны многими этиологическими факторами (инфекционные заболевания, тератогены, нарушения метаболизма и ишемия миокарда).
ЭПИДЕМИОЛОГИЯ
Врождённые кардиомиопатии встречаются относительно редко — не более, чем в 1% случаев всех врождённых пороков сердца.
ЭТИОЛОГИЯ
Врождённые кардиомиопатии проявляются кардиомегалией и гипертрофией стенок желудочков, а также снижением их сократительной функции. Клинические проявления колеблются от стёртых форм, которые становятся заметными только в детском возрасте, до приводящих к застойной сердечной недостаточности и смерти в пренатальном периоде.
ТЕТРАДА ФАЛЛО
Сложный порок развития, включающий несколько сердечных аномалий:
●дефект межжелудочковой перегородки;
●декстрапозицию аорты;
●обструкцию выходного отдела лёгочной артерии;
●гипертрофию правого желудочка.
КОД ПО МКБ-10
Q21.8 Тетрада Фалло.
ЭПИДЕМИОЛОГИЯ
Живорождённых с врождёнными пороками сердца тетраду Фалло выявляют в 4–11% случаев — это одна из наиболее частых форм врождённых пороков сердца у новорождённых с цианозом.
ДИАГНОСТИКА
При выявлении этой патологии необходимо комплексное обследование и пренатальное консультирование. Частота сочетанных экстракардиальных пороков, составляет 30–45%, хромосомные аномалии обнаруживают у 10–22% плодов.
ОБЩИЙ АРТЕРИАЛЬНЫЙ СТВОЛ КОД ПО МКБ-10
Q20.0 Общий артериальный ствол.
ЭПИДЕМИОЛОГИЯ
Доля общего артериального ствола составляет 1–4% всех врождённых пороков сердца. В 20% случаев эта аномалия сочетается с экстракардиальными пороками и хромосомными нарушениями.
КЛАССИФИКАЦИЯ
В зависимости от морфологии лёгочной артерии выделяют три типа.
●Тип I характеризуется наличием общей лёгочной артерии, которая даёт начало правой и левой ветвям приблизительно на одном уровне.
●При II и III типах общего артериального ствола ветви лёгочных артерий выходят раздельно на различных уровнях.
ЭТИОЛОГИЯ
При общем артериальном стволе из сердца выходит один артериальный сосуд, дающий начало системному, коронарному и лёгочному кровообращению. Этому пороку сердца всегда сопутствует дефект межжелудочковой перегородки, над которым располагается единственный выходящий сосуд.
ТАКТИКА ВРАЧА
До недавнего времени развитие общего артериального ствола сопровождалось достаточно высокой естественной летальностью (до достижения возраста 6 мес умирало 50–70% детей), в последние годы кардиохирурги достигли существенных успехов в лечении данного порока.
ТРАНСПОЗИЦИЯ ГЛАВНЫХ АРТЕРИЙ ЭПИДЕМИОЛОГИЯ
На долю полной формы транспозиции главных артерий приходится 5–7% от всех врождённых пороков развития сердца. Это наиболее частый среди пороков синего типа и вторым по частоте врождённым пороком сердца, обнаруживаемым в младенческом возрасте. Корригированная форма составляет в среднем 1,5% от всех врождённых пороков сердца.
Из живорождённых с транспозицией главных артерий выживает 31–50% детей.
КЛАССИФИКАЦИЯ
В зависимости от нарушения конусостволовой сегментации взаиморасположение аорты и лёгочной артерии может быть различным, в большинстве случаев аорта находится спереди и справа от лёгочной артерии.
Выделяют две формы транспозиции главных артерий: полную и корригированную.
●При полной форме аорта выходит из правого желудочка, а лёгочная артерия — из левого желудочка.
●Корригированная форма транспозиции главных артерий характеризуется предсердно-желудочковой и желудочковоартериальной дискордантностью с сохранением физиологического кровотока.
ЭТИОЛОГИЯ
Причина транспозиции главных артерий — недостаточная спирализация аортопульмональной перегородки. Хромосомные аномалии и сопутствующие экстракардиальные пороки диагностируют редко.
ТАКТИКА ВРАЧА
Специфической тактики ведения при обнаружении транспозиции главных артерий в пренатальном периоде не существует. В последние годы кардиохирурги достигли существенных успехов в лечении этого порока.
КОАРКТАЦИЯ АОРТЫ
Коарктация аорты представляет собой сужение ее просвета.
КОД ПО МКБ-10
Q25.1 Коарктация аорты.
ЭПИДЕМИОЛОГИЯ
Коарктация аорты составляет 6–8% от всех врождённых пороков сердца — четвёртая по частоте причина критического состояния новорождённых при аномалиях сердечно-сосудистой системы.
КЛАССИФИКАЦИЯ
Коарктация в большинстве случаев формируется в нисходящей части аорты вблизи места впадения артериального протока. Степень выраженности коарктации может быть разной: от минимальной, не требующей коррекции, до критической, при которой необходима операция в первые дни жизни.
ЭТИОЛОГИЯ
Выраженную коарктацию аорты относят к летальным состояниям. Умеренная коарктация может приводить к гипертрофии миокарда левого желудочка, сердечной недостаточности, системной гипертензии и заболеваниям сосудов головного мозга и коронарных артерий.
ТАКТИКА ВРАЧА
Хирургическое лечение обычно заключается в удалении сегмента коарктации и наложении анастомоза конец в конец. Смертность варьирует от 0 до 24%, составляя в среднем 12%.
ВРОЖДЁННЫЙ СТЕНОЗ АОРТЫ ЭПИДЕМИОЛОГИЯ
На долю врождённого стеноза аорты приходится от 3 до 6% всех врождённых пороков.
ЭТИОЛОГИЯ
При выраженном аортальном стенозе возникает существенное превалирование её диаметра (постстенотическое расширение) над диаметром лёгочной артерии, расширение левого предсердия, митральная регургитация и левоправый шунт через овальное окно. Часто отмечают аномальное развитие створок аортального клапана.
Исход при стенозе аорты, в первую очередь, зависит от степени тяжести обструкции выходного тракта левого желудочка. Смертность на первом году жизни не превышает 10%, исключение составляют только случаи критического аортального стеноза, прогноз при которых неблагоприятный, особенно при обнаружении в пренатальном периоде признаков застойной сердечной недостаточности.
СТЕНОЗ И АТРЕЗИЯ ЛЁГОЧНОЙ АРТЕРИИ
Эти пороки относят к обструктивным поражениям выходного отдела правого желудочка.
КОД ПО МКБ-10
Q25.6 Стеноз и атрезия лёгочной артерии.
ЭПИДЕМИОЛОГИЯ
Одни из наиболее частых врождённых пороков сердца — частота их развития составляет до 20% от общего числа.
ДИАГНОСТИКА
По мнению многих исследователей, слабо и умеренно выраженные формы стеноза и атрезии лёгочной артерии обычно остаются не диагностированными в пренатальном периоде при проведении скринингового ультразвукового обследования, поскольку эхографическая картина на четырёхкамерном срезе и срезах главных артерии практически не изменяется при этих формах порока. При данном пороке часто обнаруживают постстенотическое расширение лёгочной артерии, размеры правого желудочка могут быть нормальными, увеличенными или уменьшенными в сочетании с гипертрофированными стенками.
Особое значение имеет пренатальная диагностика критического стеноза и атрезии лёгочной артерии, так как прогноз в этих случаях не столь оптимистичный, как при слабо и умеренно выраженных формах. При атрезии лёгочная артерия имеет маленький диаметр, нередко нетипично расположена. При интактной межжелудочковой перегородке миокард правого желудочка часто гипертрофирован, правое предсердие дилатировано. При атрезии лёгочной артерии кровь в неё поступает из аорты через артериальный проток, прямой поток крови из правого желудочка в лёгочную артерию отсутствует.
ДВОЙНОЙ ВЫХОД ГЛАВНЫХ АРТЕРИЙ ИЗ ПРАВОГО ЖЕЛУДОЧКА
Этот порок характеризуется совместным выходом лёгочной артерии и аорты из правого желудочка.
ЭПИДЕМИОЛОГИЯ
На долю двойного выхода главных артерий из правого желудочка приходится около 1% от всех врождённых пороков сердца. Этот порок в изолированном виде, как правило, не встречается. С ним часто сочетаются дефекты межжелудочковой перегородки, стеноз лёгочной артерии, коарктация аорты и аномалии атриовентрикулярных клапанов.
ДИАГНОСТИКА
Многие исследователи считают диагностической дилеммой пренатальное обнаружение двойного выхода главных артерий из правого желудочка, что обусловлено как разнообразием анатомических вариантов порока, так и сходством эхографической картины аномалии с тетрадой Фалло и выраженной декстрапозицией аорты. Новые возможности в точной идентификации двойного выхода главных сосудов из правого желудочка у плода после обнаружения аномальных изображений различных срезов сердца и главных артерий открывает технология, которая позволяет проводить детальный (при необходимости даже ретроспективный) анализ изображений, включая режимы и, в любой плоскости.
АНОМАЛИИ РАЗВИТИЯ ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА
Различные врождённые аномалии желудочно-кишечного тракта и передней брюшной стенки встречаются с частотой около 20–25 случаев на 1000 новорождённых или 4 случая на 100 перинатальных вскрытий.
АТРЕЗИЯ ПИЩЕВОДА
Атрезия пищевода представляет собой отсутствие его сегмента и в подавляющем большинстве случаев сопровождается развитием трахеопищеводной фистулы.
КОД ПО МКБ-10
Q39 Атрезия пищевода.
ЭПИДЕМИОЛОГИЯ
Частота атрезии пищевода составляет в среднем 2–3 случая на 10 000 живорождённых.
КЛАССИФИКАЦИЯ
Описано 5 типов атрезии пищевода:
●изолированная атрезия (4%);
●атрезия со свищом, когда верхний сегмент слепой, а нижний соединён с трахеей (90%);
●атрезия со свищом, когда верхний сегмент соединён с трахеей, а нижний слепой (1%);
●атрезия, при которой оба сегмента пищевода соединены с трахеей в одном месте (4%);
●атрезия, когда оба сегмента пищевода соединены с трахеей в разных местах (1%).
ДИАГНОСТИКА
Пренатальная эхографическая диагностика атрезии пищевода основана только на косвенных признаках:
●отсутствие эхографического изображения желудка или маленькие его размеры;
●многоводие, связанное со снижением оборота околоплодных вод вследствие непроходимости пищевода.
Атрезия пищевода в более половины случаев (63,2–72%) сочетается с другими пороками, среди которых наиболее часто регистрируют врождённые пороки сердца, аномалии желудочно-кишечного тракта, мочеполовой системы и опорно-двигательного аппарата. Также нередки хромосомные аномалии, из которых доминирует трисомия 18.
ТАКТИКА ВРАЧА
Атрезия пищевода — прогностически неблагоприятный врождённый порок развития.
АТРЕЗИЯ ДВЕНАДЦАТИПЕРСТНОЙ КИШКИ КОД ПО МКБ-10
Q41.0 Атрезия двенадцатиперстной кишки.
ЭПИДЕМИОЛОГИЯ
Этот порок — наиболее частая причина врождённой непроходимости тонкого кишечника. Частота этой патологии составляет 1:10 000 живорождённых.
Атрезия двенадцатиперстной кишки — изолированный порок лишь у 30–52% плодов. Среди сочетанных аномалий наиболее часто отмечают врождённые пороки сердца и мочевыделительной системы, мальротацию и другие аномалии желудочно-кишечного тракта, дефекты позвоночника, высока частота сочетаний с хромосомными аберрациями.
ДИАГНОСТИКА
Ультразвуковая диагностика порока основана на одновременной визуализации расширенного желудка и двенадцатиперстной кишки (симптом двух пузырей, «double-bubble»). Во всех случаях атрезии двенадцатиперстной кишки диагностируют многоводие.
ТАКТИКА ВРАЧА
Показано пренатальное кариотипирование. В случаях, когда родители принимают решение о продлении беременности, при выраженном многоводии целесообразно проведение лечебного амниоцентеза.
АНОМАЛИИ ЖЕЛУДКА КОД ПО МКБ-10
Q40.2 Аномалии желудка.
ЭПИДЕМИОЛОГИЯ
Аномалии желудка относят к разряду чрезвычайно редких, составляющих менее 1% среди всех пороков развития желудочно-кишечного тракта.
КЛАССИФИКАЦИЯ
В своей структуре аномалии желудка многочисленны и разнообразны. К наиболее часто встречающимся и наиболее важным в клиническом отношении относят:
●агенезию и атрезию желудка (атрезия пилорического отдела);
●гипоплазию желудка (микрогастрия врождённая);
●стеноз привратника желудка гипертрофический врождённый (пилоростеноз гипертрофический);
●удвоение желудка (желудок двойной).
АТРЕЗИИ И СТЕНОЗЫ ТОЩЕЙ И ПОДВЗДОШНОЙ КИШКИ КОД ПО МКБ-10
Q41 Атрезии и стенозы тощей и подвздошной кишок.
ЭПИДЕМИОЛОГИЯ
Пороки этой группы встречаются с частотой 0,07 на 1000 живорождённых, причём атрезии в 20 раз чаще, чем стенозы.
ЭТИОЛОГИЯ
Чаще поражается проксимальная часть тощей и дистальный отдел подвздошной кишки. Около 15% атрезий носит множественный характер. Нередко эта патология сочетается с нарушением поворота кишечника и муковисцидозом.
ДИАГНОСТИКА
Многоводие — один из первых симптомов атрезии тонкого кишечника, сочетающийся с этим пороком развития после 24 нед беременности в 75% случаев.
В подавляющем большинстве случаев атрезия тонкой кишки диагностируется в конце II — начале III триместров беременности.
Хромосомные аберрации встречаются редко, поэтому от пренатального кариотипирования можно отказаться.
АТРЕЗИИ И СТЕНОЗЫ ТОЛСТОЙ КИШКИ
КОД ПО МКБ-10
Q42 Атрезии и стенозы толстой кишки.
ЭПИДЕМИОЛОГИЯ
Составляют 1,8–10% всех атрезий и стенозов кишечника, их диагностируют с частотой 1 случай на 20 000 живорождённых.
КЛАССИФИКАЦИЯ
Различают полную и частичную окклюзию толстой кишки, которая может быть обусловлена наличием внутренней диафрагмы (30%), разделением проксимального и дистального отделов кишечника фиброзными тяжами (20%) или полной их сепарацией (50%).
ДИАГНОСТИКА
Атрезию толстой кишки можно заподозрить при обнаружении расширенных петель толстого кишечника в III триместре, однако большинство случаев атрезии толстой кишки антенатально не диагностируют. Риск хромосомных аномалий при атрезии толстой кишки не выше популяционного.
БОЛЕЗНЬ ГИРШПРУНГА
Синонимы
Врождённый аганглиоз, colon aganglionosis, megacolon.
КОД ПО МКБ-10
Q43.1 Болезнь Гиршпрунга.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости — 0,2 на 1000 новорождённых. Чаще болеют мальчики (75–80%).
КЛАССИФИКАЦИЯ
В зависимости от локализации аганглиозных участков выделяют ректальную (21,9%), ректосигмоидальную (69,2%), субтотальную (3,2%), тотальную (0,6%) и сегментарную (5,1%) формы болезни Гиршпрунга.
ЭТИОЛОГИЯ
Причина заболевания — отсутствие нейронов межмышечного (ауэрбаховского) сплетения нижнего отрезка сигмовидной и прямой кишки. Вследствие сохранности подслизистого (мейснеровского) сплетения аганглионарный участок кишки спастически сокращён, выше него происходит растяжение кишки меконием с развитием последующей компенсаторной гипертрофии мышечной оболочки. В растянутом отрезке кишки иногда возникают изъязвления, развивается непроходимость. Этиология порока мультифакториальная.
ДИАГНОСТИКА
Выявить болезни Гиршпрунга в пренатальном периоде проблематично.
АТРЕЗИЯ АНУСА КОД ПО МКБ-10
Q42.1 Атрезия ануса.
ЭПИДЕМИОЛОГИЯ
Встречается с частотой 2 случая на 10 000 живорождённых, причём у девочек в 2 раза реже, чем у мальчиков.
ДИАГНОСТИКА
В большинстве случаев специфические пренатальные эхографические изменения отсутствуют.
Атрезия ануса сочетается с другими пороками, различными хромосомными дефектами и наследственными синдромами.
МЕКОНИЕВЫЙ ПЕРИТОНИТ
Мекониевый перитонит — асептическая воспалительная реакция, возникающая в ответ на перфорацию кишечника плода и выход мекония в брюшную полость.
ЭПИДЕМИОЛОГИЯ
Частота заболевания составляет 0,29–0,33 на 1000 живорождённых.
КЛАССИФИКАЦИЯ
В зависимости от степени распространения воспалительного процесса выделяют три типа мекониевого перитонита.
●Генерализованный тип характеризуется наличием диффузного перитонеального фиброзного уплотнения и отложением кальция, меконий распространяется по всей перитонеальной полости.
●При фиброадгезивном (локализованном) варианте формируется плотное образование с включениями кальция, которое закрывает перфорационное отверстие, приводя к непроходимости.
●Если перфорационное отверстие не закрывается полностью, формируются толстостенные кисты (энтерокистомы) за счёт адгезии проксимальных петель кишечника к перфорационному отверстию (кистозный тип).
ЭТИОЛОГИЯ
Мекониевый перитонит может быть обусловлен перфорацией кишечника в результате его непроходимости вследствие стриктуры, атрезии, заворота, инвагинации и мекониевого илеуса при кистозном фиброзе. Возможные причины перфорации кишечника:
●сосудистая недостаточность кишечника;
●гидрометрокольпос;
●аппендицит плода;
●внутриутробное инфицирование (цитомегаловирус, гепатит А, В, парвовирус В19).
ДИАГНОСТИКА
При генерализованном типе обычно обнаруживают многоводие, асцит, множество кальцинатов в брюшной полости. Фиброадгезивный тип характеризуется расширением петель кишечника.
ТАКТИКА ВРАЧА
Пренатальная тактика в случае обнаружения мекониевого перитонита до конца не определена.
АНОМАЛИИ РАЗВИТИЯ ОПОРНО-ДВИГАТЕЛЬНОЙ СИСТЕМЫ
Врождённые пороки развития опорно-двигательной системы плода — многочисленная гетерогенная группа аномалий (описано более 270 форм) различных по этиологии, патогенезу и клиническим проявлениям. Аномалии этой группы встречаются значительно реже других пороков — в среднем в 2–3 случаях на 10 000 новорождённых.
АХОНДРОПЛАЗИЯ
Ахондроплазия — одна из редких скелетных дисплазий, совместимая с жизнью и практически не требующая социальной адаптации, однако гомозиготная форма аномалии летальна и проявляет себя максимально выраженным укорочением конечностей и узкой грудной клеткой.
Синонимы Хондродистрофия.
КОД ПО МКБ-10
Q77.4 Ахондроплазия.
ЭПИДЕМИОЛОГИЯ
Встречается с частотой 1 случай на 10 000–26 000 новорождённых.
ЭТИОЛОГИЯ
Ахондроплазия — скелетная дисплазия, чаще спорадического характера, хотя и может быть аутосомно-доминантным заболеванием.
ДИАГНОСТИКА
Характерные признаки гетерозиготной ахондроплазии:
●проксимальное укорочение конечностей;
●поясничный лордоз;
●короткие пальцы, расположенные в форме трезубца;
●макроцефалия с выступающим лбом и запавшей переносицей. Отличительные признаки данного заболевания от других скелетных дисплазий:
●макроцефалия;
●макрофтальмия;
●гипертелоризм;
●выступающий лоб и затылок;
●седловидный нос.
Трубчатые кости, как правило, сохраняют правильную форму, деформируются лишь в 25–30% случаев. Укорочение длинных трубчатых костей происходит преимущественно по ризомелическому типу, однако нередко развивается и симметричное укорочение. Грудная клетка и живот плода соответствуют нормативным значениям для данного срока беременности. При ахондроплазии часто отмечают многоводие.
Пренатальный диагноз ахондроплазии можно поставить не ранее 22 нед. |
|
|
ТАНАТОФОРНАЯ ДИСПЛАЗИЯ |
|
|
Синонимы |
|
|
Танатофорная карликовость. |
|
|
ЭПИДЕМИОЛОГИЯ |
|
|
Одна из наиболее распространённых скелетных дисплазий, |
частота которой составляет |
0,2–0,7 на |
10 000 родившихся. |
|
|
ЭТИОЛОГИЯ |
|
|
Танатофорная дисплазия — летальная остеохондродисплазия, в |
основе которой лежит нарушение |
процесса |
эндохондрального окостенения.
ДИАГНОСТИКА
Основные признаки:
● выраженное укорочение трубчатых костей по ризомелическому типу; ● узкая грудная клетка при нормальной длине туловища.
● макроцефалия; ● выступающий лоб;
● изменённая форма черепа в виде трилистника.
В ряду сочетанных аномалий при танатофорной дисплазия наиболее часто встречается гидроцефалия, пороки сердца и мочевыделительной системы. В большинстве случаев в антенатальном периоде эта аномалия развития сопровождается многоводием.
Пренатальная диагностика танатофорной дисплазии возможна с середины II триместра беременности.
НЕСОВЕРШЕННЫЙ ОСТЕОГЕНЕЗ
Это группа наследственных болезней, включающая в себя такие синдромы, как Элерса–Данло и Марфана, для которых характерны генерализованная остеопения и хрупкость костей.
Синонимы
Синдром Van der Hoeve, синдром Eddowe, болезнь Лобштейна–Экмана, болезнь Порака–Дюранта, болезнь «стеклянных костей», синдром «голубых склер».
КОД ПО МКБ-10
Q78.0 Несовершенный остеогенез.
КЛАССИФИКАЦИЯ
Несовершенный остеогенез делят на типы и подтипы, которые различаются по патогенезу и клиническим проявлениям.
● Несовершенный остеогенез II типа (врождённая хрупкость костей) относится к летальным синдромам и характеризуется низкими весовыми и ростовыми показателями при рождении, рентгенологическими признаками множественных переломов трубчатых костей и рёбер, возникших внутриутробно. Для этой формы порока характерны нарушение минерализации костей черепа, чёткообразная деформация рёбер, выраженное укорочение трубчатых костей и их деформация.
ЭТИОЛОГИЯ
В основе заболевания лежит генетический дефект мезенхимальной ткани, проявляющийся нарушением продукции коллагена, что приводит к неполноценности хрящевой и костной ткани, зубов, связочного аппарата. Все типы
несовершенного остеогенеза обусловлены точечными мутациями генов, кодирующих цепи про-α1- и про-α2-коллагена I типа.
ДИАГНОСТИКА
Потенциально диагностируемый в перинатальном периоде — несовершенный остеогенез II типа. В отличие от целого ряда форм скелетных дисплазий, несовершенный остеогенез II типа можно выявить при скрининговом ультразвуковом исследовании во II триместре гестации.
АХОНДРОГЕНЕЗ
Это заболевание скелета, которое по Международной классификации скелетных дисплазий относят к летальным остеохондродисплазиям.
КОД ПО МКБ-10
Q77.0 Ахондрогенез.
ЭПИДЕМИОЛОГИЯ
По встречаемости среди всех врождённых пороков развития опорно-двигательной системы ахондрогенез стоит на II месте после танатофорной дисплазии (0,23:10 000).
КЛАССИФИКАЦИЯ
По клиническим, радиологическим и гистологическим особенностям выделяют два типа ахондрогенеза.
●Ахондрогенез I типа характеризуется выраженной макроцефалией, седловидным носом, нависающим лбом, отсутствием гипертелоризма. Укорочение трубчатых костей конечностей составляет 10–14 нед от должных значений, кисти и стопы с трудом поддаются ультразвуковой идентификации, двигательная активность плода весьма ослаблена или отсутствует. Во всех случаях ахондрогенез I типа сопровождается выраженным многоводием.
●Ахондрогенез II типа отличается менее выраженным укорочением конечностей (на 5–8 нед) и меньшим нарушением
окостенения костей свода черепа и позвоночника, переломы рёбер отсутствуют. Отличие ахондрогенеза II типа от ахондроплазии заключается в размерах грудной клетки и туловища, которые при ахондроплазии всегда имеют нормальные размеры.
ЭТИОЛОГИЯ
В основе этого заболевания лежит генетически детерминированное нарушение продукции хрящевой ткани хондроцитами, что приводит к нарушению оссификации.
ДИАГНОСТИКА
Основные признаки ахондрогенеза можно выявить с помощью эхографии во II триместре беременности. Ведущие признаки ахондрогенеза:
●выраженное укорочение трубчатых костей по типу микромелии, их утолщение;
●макроцефалия;
●укорочение туловища.
Главное отличие ахондрогенеза от рассмотренной выше танатофорной дисплазии состоит в нарушении оссификации позвоночника.
КАМПОМЕЛИЧЕСКАЯ ДИСПЛАЗИЯ
Кампомелическая дисплазия — летальный аутосомно-рецессивный синдром с частотой встречаемости 1:200 000.
ДИАГНОСТИКА
Для кампомелической дисплазии характерны:
●укорочение и искривление трубчатых костей (преимущественно ног);
●узкая (колоколообразная) грудная клетка;
●гипопластичные лопатки;
●большой череп с диспропорционально маленьким лицом и лицевыми дизморфиями.
ДИАСТРОФИЧЕСКАЯ ДИСПЛАЗИЯ
Это аутосомно-рецессивно наследуемая системная скелетная дисплазия, характеризующаяся укорочением конечностей, косолапостью, деформацией кистей и стоп, множественными сгибательными контрактурами и сколиозом. Патогномоничным признаком заболевания является характерное отведение большого пальца кистей и стоп.
СИНДРОМ КОРОТКИХ РЁБЕР
Синонимы
Полидактилии (I, II, III типа).
КОД ПО МКБ-10
Q77.2 Синдром коротких рёбер ЭТИОЛОГИЯ
Синдром коротких рёбер — группа летальных дисплазий с аутосомно-рецессивным типом наследования, для которой характерны короткие рёбра, узкая грудная клетка, укороченные конечности и постаксиальная полидактилия.
ВРОЖДЁННЫЕ АМПУТАЦИИ КОНЕЧНОСТЕЙ ПЛОДА ЭПИДЕМИОЛОГИЯ
Врождённые ампутации конечностей плода встречаются значительно чаще, чем описанные выше системные дисплазии скелета. Их частота 0,54–0,59 на 1000 рождений. При включении в расчёт мертворождённых и беременностей, прерванных по медицинским показаниям, этот показатель возрастает до 0,69 на 1000.
Около половины всех случаев врожденных ампутаций — изолированное поражение одной конечности. Остальные 50% — множественные редукционные поражения, представленные составной частью сложных симптомокомплексов аномалий.
ЭТИОЛОГИЯ
Причины врождённых ампутаций разнообразны, значительная часть (30%) приходится на генные мутации и патологию хромосом, в 4% наблюдений причина аномалий — тератогенное воздействие.
Ампутационные пороки конечностей могут входить в состав ADAM-комплекса (amniotic deformations, adhesions, malitation), включающего адгезию амниона (амниотичеких перетяжек), деформации, ампутации и мутиляции плода (самопроизвольные отторжения некротизированных частей тела или органа).
ТАКТИКА ВРАЧА
Врождённые ампутации конечностей — совместимая с жизнью патология, но приводящая к тяжёлой инвалидности.
КОСОЛАПОСТЬ
Косолапость — один из частых видов поражения опорно-двигательной системы.
ЭПИДЕМИОЛОГИЯ
В пренатальном периоде она встречается с частотой 1 случай на 250 беременностей, в постнатальном периоде — 1– 3:1000, при этом 1/3 составляют изолированные случаи.
ЭТИОЛОГИЯ
Причины косолапости разнообразны:
●неудобное внутриутробное положение стопы;
●наличие структурных изменений в голеностопном суставе и др.
Постнатальные исследования показали, что из всех живорождённых с изолированной косолапостью 90% приходится на структурные аномалии суставов и лишь 10% — на функциональные.
Косолапость может быть изолированной, а также составной частью более 300 генетических синдромов, сочетаться с аномалиями других органов и систем и быть проявлением хромосомных аномалий. Пороки развития других органов встречается почти в 70% наблюдений. Хромосомные аномалии в среднем диагностируют у 15% плодов.
ДИАГНОСТИКА
Косолапость легко диагностировать при рутинной эхографии.
СИНДРОМ РАСЩЕПЛЕНИЯ КИСТЕЙ И СТОП
синонимы Синдром Гертвига–Вейерса
ЭПИДЕМИОЛОГИЯ
Частота возникновения этого синдрома у новорождённых очень низкая — 1:90 000–150 000.
ЭТИОЛОГИЯ
Синдром расщепления кистей и стоп — аутосомно-доминантный порок, при котором кисть и стопа в виде клешни формируются за счёт отсутствия фаланг средних пальцев и костей запястья.
ЭКТРОДАКТИЛИЯ
КЛАССИФИКАЦИЯ
Различают монодактилию (1 палец), дидактилию (2 пальца), тридактилию (3 пальца) и тетрадактилию (4 пальца). Чаще всего встречается дидактилия («клешня рака»), причём иногда расщепление между пальцами распространяется на область запястья и пястные кости («клешня омара»).
ЭТИОЛОГИЯ
Эктродактилия — аномалия развития, проявляющаяся в отсутствии пальцев рук и ног. Эктродактилия может быть следствием недоразвития зачатков пальцев или следствием слияния пальцев, ведущего к уменьшению числа их и именуемого синдактилией.
ДЕФОРМАЦИЯ КИСТЕЙ
Синонимы Косорукость.
КЛАССИФИКАЦИЯ
Деформации кистей подразделяют на лучевые и локтевые.
При лучевой косорукости большой палец гипоплазирован или отсутствует, лучевая кость отсутствует. Лучевая косорукость часто входит в состав различных синдромов.
Локтевая косорукость — более редкое состояние, характеризующееся деформацией локтевой кости вплоть до её отсутствия. Эта патология чаще всего встречается в изолированном виде.
ЭТИОЛОГИЯ
Деформация кистей нередко связана с хромосомными аномалиями (синдром Эдвардса), гематологическими заболеваниями [панцитопения Фанкони, синдром TAR (Thrombocytopenia–Absent radius)] или генетическими синдромами (синдром Холт–Орама).
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Алгоритмы пренатальной диагностики. Учебное пособие / Под ред. М.В. Медведева. — М.: Реальное время, 2005. —
С. 27.
Медведев М.В., Юдина Е.В. Дифференциальная пренатальная ультразвуковая диагностика. — М.: Реальное Время, 2004.
Пренатальная эхография / Под ред. М.В. Медведева, 1-е изд. — М.: Реальное время, 2005.
Снайдерс Р.Дж.М., Николаидес К.Х. Ультразвуковые маркёры хромосомных дефектов плода. — М.: Видар, 1997. Флейшер А., Меннинг Ф., Дженти Ф. et al. Эхография в акушерстве и гинекологии. Часть I: пер. с англ. / Под ред. Е.В. Фёдоровой, А.Д. Липмана. — М., 2005.
Эхокардиография плода / Под ред. М.В. Медведева. — М.: РАВУЗДПГ, Реальное Время, 2000.
Юдина Е.В. Ультразвуковые пренатальные маркёры хромосомных аномалий во II триместре беременности: Дисс. … докт. мед. наук. — М., 2003.
Rilbsinghani A., Yankowitz J., Kanis A. et al. Antenatal sonographic diagnosis of club foot with particular attention to the implications and outcomes of isolated club foot // Ultrasound Obstet. Gynecol. — 1998. — Vol. 11 — P. 103–106.