

7 семестр / Основы_физич_химии_Теория_и_задачи_Еремин_и_др_2005_480с
.pdf14Г л а в а 1. Основы химической термодинамики
•необратимые, или неравновесные, когда в результате процесса невозможно возвратить и систему, и ее окружение к первоначальному состоянию.
Входе процесса некоторые термодинамические переменные могут быть зафиксированы. В частности, различают изотермический (T = const), изохорный (V = const), изобарный (p = const) и адиабатический (Q = 0, δQ = 0) процессы.
Термодинамические функции разделяют на:
•функции состояния, которые зависят только от состояния системы и не зависят от пути, по которому это состояние получено;
•функции перехода, значение которых зависит от пути, по которому происходит изменение системы.
Примеры функций состояния: энергия U, энтальпия H, энергия Гельмгольца F, энергия Гиббса G, энтропия S. Термодинамические переменные – объем V, давление p, температуру T – также можно считать функциями состояния, т.к. они однозначно характеризуют состояние системы. Примеры функций перехода: теплота Q и работа W.
Функции состояния характеризуются следующими свойствами:
•бесконечно малое изменение функции f является полным дифференциалом (обозначается df);
•изменение функции при переходе из состояния 1 в состояние 2 оп-
2
ределяется только этими состояниями: ∫ df = f 2 − f1 ;
1
•в результате любого циклического процесса функция состояния не изменяется: ∫vdf = 0 .
Существует несколько способов аксиоматического построения термодинамики. В настоящем издании мы исходим из того, что выводы и соотношения термодинамики можно сформулировать на основе двух постулатов (исходных положений) и трех законов (начал).
Первое исходное положение, или основной постулат термодинамики:
Любая изолированная система с течением времени приходит в равновесное состояние и самопроизвольно не может из него выйти.
Это положение ограничивает размер систем, которые описывает термодинамика. Оно не выполняется для систем астрономического масштаба и микроскопических систем с малым числом частиц. Системы галактического размера самопроизвольно не приходят в состояние равновесия благодаря дальнодействующим гравитационным силам. Микроскопические системы могут самопроизвольно выходить из состояния равновесия; это явление называют флуктуациями. В статисти-
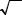
Г л а в а 1. Основы химической термодинамики |
15 |
ческой физике показано, что относительная величина флуктуаций термодинамических величин имеет порядок 1/ N , где N – число частиц в системе. Если считать, что относительные значения меньше 10–9 невозможно обнаружить экспериментально, то нижний предел для числа частиц в термодинамической системе составляет 1018.
Самопроизвольный переход системы из неравновесного состояния в равновесное называют релаксацией. Основной постулат термодинамики ничего не говорит о времени релаксации, он утверждает, что равновесное состояние системы будет обязательно достигнуто, но длительность такого процесса никак не определена. В классической равновесной тер-
модинамике вообще нет понятия времени.
Для того, чтобы использовать термодинамику для анализа реальных процессов, необходимо выработать некоторые практические критерии, по которым можно было бы судить о завершенности процесса, т.е. достижении равновесного состояния. Состояние системы можно считать равновесным, если текущее значение переменной отличается от равновесного на величину, меньшую, чем ошибка, с которой эта переменная измеряется. Релаксационный процесс можно считать закончившимся, если наблюдаемое свойство системы остается неизменным в течение времени, сопоставимого со временем релаксации по этой переменной. Так как в системе одновременно могут протекать несколько процессов, при рассмотрении условий достижения равновесия надо сопоставлять времена релаксации по разным переменным. Очень часто неравновесная в целом система оказывается равновесной по отношению к процессам с малыми временами релаксации, и их термодинамическое описание оказывается вполне корректным.
Второе исходное положение, или нулевой закон термодинамики описывает свойства систем, находящихся в состоянии теплового равновесия:
Если система А находится в тепловом равновесии с системой В, а та, в свою очередь, находится в равновесии с системой С, то системы А и С также находятся в тепловом равновесии.
Второй постулат говорит о существовании особой интенсивной переменной, характеризующей состояние теплового равновесия и называемой температурой. Системы, находящиеся в тепловом равновесии, имеют одинаковую температуру. Таким образом, нулевой закон – это постулат о существовании температуры. Транзитивностью обладает не только тепловое, но и любое другое равновесие (механическое, диффузионное и т.п.), но в термодинамике постулируется только термическое равновесие, а выравнивание всех остальных интенсивных переменных на контрольной поверхности является следствием этого постулата и второго закона термодинамики.
16 |
Г л а в а 1. Основы химической термодинамики |
Уравнения состояния
(1.1)
(1.2)
(1.3)
Из постулатов термодинамики следует, что при равновесии внутренние переменные термодинамической системы являются функциями внешних переменных и температуры. Например, если система содержит K компонентов, занимает объем V и имеет температуру T, то при равновесии любые термодинамические характеристики этой системы, такие как количества и концентрации образовавшихся соединений, число фаз, давление, теплоемкость, коэффициент термического расширения и другие являются функциями не более, чем (K + 2) независимых переменных. Если же система закрыта, т.е. не может обмениваться веществом с окружением, то для описания ее свойств достаточно двух независимых переменных. Отсюда следует вывод о существовании уравнения состояния термодинамической системы, связывающего внутренние переменные с внешними переменными и температурой или внутренней энергией. В общем случае уравнение состояния имеет вид:
f (a, b,T ) = 0 или a = a(b,T ) ,
где a – совокупность внутренних параметров, b – совокупность внешних параметров, T – температура.
Если внутренним параметром является давление, а внешним – объем, то уравнение состояния
p = p(V , n,T )
называют термическим. Если внутренним параметром является энергия, а внешним – объем, то уравнение состояния
U =U (V , n,T )
называют калорическим.
Количество независимых уравнений состояния равняется вариантности системы, т.е. числу независимых переменных, достаточных для описания термодинамического состояния равновесной системы (оно на единицу больше числа внешних переменных).
В случае закрытой системы в отсутствие внешних полей и поверхностных эффектов число внешних переменных равно 1 (V), соответственно, число уравнений состояния равно 2. Если открытая система содержит K компонентов и может изменять объем, то число внешних переменных составляет K + 1, а число уравнений состояния равно
K + 2.
Если известны термическое и калорическое уравнения состояния, то аппарат термодинамики позволяет определить все термодинамические свойства системы, т.е. получить ее полное термодинамическое описа-

|
|
|
Г л а в а 1. Основы химической термодинамики |
|
|
|
17 |
||||||||
ние. Сами уравнения состояния нельзя вывести методами классической |
|
|
|||||||||||||
термодинамики, но их можно определить экспериментально. |
|
|
|
|
|||||||||||
Самым простым уравнением состояния описывается идеальный газ – |
|
|
|||||||||||||
совокупность невзаимодействующих частиц точечного размера: |
|
|
|
||||||||||||
|
|
|
|
|
|
|
pV = nRT, |
|
|
|
|
|
(1.4.а) |
|
|
где R – универсальная газовая постоянная, или |
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
pVm = RT, |
|
|
|
|
|
(1.4.б) |
|
|
где Vm = V/n – мольный объем газа (см. рис. 1.1). |
|
|
|
|
|
||||||||||
Реальные газы лишь прибли- |
|
|
|
|
|
|
|
||||||||
женно |
описываются |
уравнением |
|
|
|
|
|
|
|
||||||
состояния идеального газа. При вы- |
|
|
|
|
|
|
|
||||||||
соких давлениях и низких темпера- |
|
|
|
|
|
|
|
||||||||
турах (особенно когда газ близок к |
|
|
|
|
|
|
|
||||||||
конденсации) отклонения от иде- |
|
|
|
|
|
|
|
||||||||
ального поведения становятся зна- |
|
p |
|
|
|
|
|
||||||||
чительными. |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Удобной мерой неидеальности |
|
|
|
|
|
|
|
||||||||
является |
фактор |
сжимаемости |
|
|
|
|
|
|
|
||||||
Z = pVm/RT, поскольку для идеаль- |
|
|
|
|
|
|
|
||||||||
ного газа Z = 1 при любых условиях. |
|
|
|
|
|
|
|
||||||||
На рис. 1.2 представлены факторы |
|
|
|
|
|
|
|
||||||||
сжимаемости некоторых реальных |
|
|
|
V |
|
|
|
||||||||
газов как функции давления при |
Изотермы идеального газа |
|
Рис. 1.1 |
|
|||||||||||
298 К |
(пунктир |
соответствует |
иде- |
|
22,0. |
|
|
|
|
|
|||||
альному газу). Из рисунка видно, |
|
|
|
|
|
|
|||||||||
что |
при |
высоких |
давлениях |
для |
|
11,8. |
|
|
|
|
|
||||
всех |
газов Z > 1, |
т.е. их труднее |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|||||||||
сжать, чем идеальный газ, посколь- |
//RTRT |
11,6. |
|
|
N2 |
|
|
||||||||
ку в этой области преобладают силы |
1. |
|
|
|
|
|
|||||||||
межмолекулярного |
отталкивания. |
|
|
|
|
|
|||||||||
m |
|
|
|
He |
|||||||||||
m |
1,4 |
|
|
|
|||||||||||
При низких давлениях для некото- |
pV |
1. |
H2 |
|
|
|
|
||||||||
ZZ== |
|
|
|
|
|||||||||||
рых |
газов |
Z < 1, |
что |
объясняется |
1,2 |
|
|
O2 |
|
|
|||||
|
|
|
|
|
|||||||||||
преобладанием |
межмолекулярного |
|
1. |
|
|
|
|
|
|||||||
притяжения. При p → 0 эффект |
|
1,0 |
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|||||||||
межмолекулярного |
взаимодействия |
|
00,8. |
CH4 |
|
|
|
|
|||||||
исчезает, потому что длина свобод- |
|
|
|
|
|
|
|||||||||
|
0. |
|
|
|
|
|
|||||||||
ного |
|
пробега молекул |
становится |
|
200 |
|
600 |
|
1000 |
||||||
|
|
0,600 |
400 |
800 |
|||||||||||
много |
больше |
расстояния |
между |
|
|
|
p, бар |
|
|
||||||
частицами, и для всех газов Z → 1, |
|
Зависимость фактора |
|
Рис. 1.2 |
|
||||||||||
т.е. в этих условиях все газы ведут |
сжимаемости некоторых |
|
|
|
|||||||||||
себя почти идеально. |
|
|
|
газов от давления при 298 K |
|
|
|
||||||||
18Г л а в а 1. Основы химической термодинамики
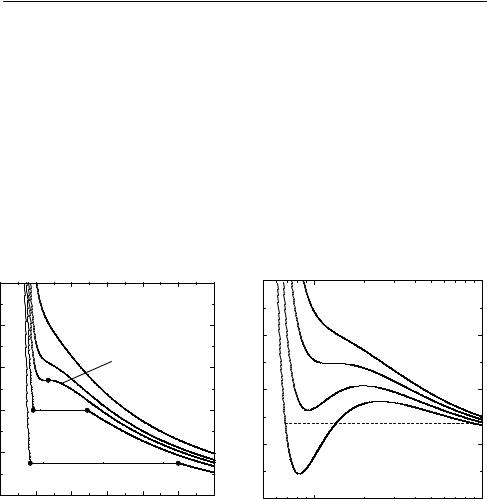
Вотличие от изотерм идеального газа (рис. 1.1), на изотермах реальных газов (рис. 1.3.а) наблюдаются горизонтальные участки, соответствующие процессу конденсации газа в жидкость. Давление, соответствующее горизонтальным участкам, когда жидкость и пар находятся в равновесии, называют давлением насыщенного пара жидкости при данной температуре. При повышении температуры длина
этих участков уменьшается, и при критической температуре Tc на изотерме наблюдается точка перегиба – критическая точка. Она ха-
рактеризуется критической температурой Tc, критическим давлением pc и критическим мольным объемом Vc. Критические константы некоторых газов приведены в приложении (табл. П-2).
а) |
|
|
|
|
|
|
|
б) |
|
|
|
|
120 |
|
|
|
|
|
|
|
150 |
|
|
|
|
|
|
60oC |
|
|
|
|
|
|
|
1.1 T |
|
|
100 |
|
|
|
|
|
|
|
|
|
c |
|
|
|
|
|
|
|
|
|
100 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
80 |
|
40oC |
31.04 oC (Tc) |
|
бар,p |
|
|
Tc |
|
|
||
|
20oC |
|
|
|
|
|
|
|
|
|
||
бар,p |
|
|
|
|
|
50 |
|
0.9 Tc |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
60 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
3 |
|
2 |
40 |
|
0oC |
|
|
|
|
|
0 |
|
0.8 Tc |
|
|
|
|
|
|
|
|
|
|
|
|
|||
20 |
0.1 |
0.2 |
0.3 |
0.4 |
0.5 |
0.6 |
|
-50 |
1 |
2 |
5 |
10 |
0.0 |
|
0.5 |
||||||||||
|
|
V, л моль−1 |
|
|
|
|
|
V, л моль−1 |
|
|
||
Рис. 1.3 |
Изотермы CO2: а) экспериментальные; |
|
б) рассчитанные по уравнению Ван-дер-Ваальса |
|
Для описания реальных газов используют более сложные уравнения состояния, в которых межмолекулярные взаимодействия учитывают с помощью эмпирических параметров, индивидуальных для каждого газа. Некоторые уравнения состояния реальных газов приведены в табл. 1.1. Известно более ста подобных уравнений, отличающихся числом параметров, степенью точности и областью применимости. Выяснилось, что ни одно из уравнений состояния, содержащих менее 5 индивидуальных постоянных, не является достаточно точным для описания реальных газов в широком диапазоне p, V, T, и все эти уравнения оказались непригодными в области конденсации газов. Из простых уравнений с двумя индивидуальными параметрами удовлетворительные результаты дают уравнения Бертло и Дитеричи
(табл. 1.1).

|
|
Таблица 1.1 |
|
|
|
|
Уравнения состояния реальных газов (для одного моля) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Уравнение |
|
Обычная форма |
|
|
|
Приведенная форма |
|
|
|
|
pc |
|
|
|
|
|
|
|
|
Tc |
|
|
Vc |
|
|
|
|
Zc |
|
||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
Ван-дер-Ваальса (1873) |
|
|
|
p = |
|
RT |
|
|
|
− |
|
a |
|
|
|
|
|
|
|
pr |
= |
|
8Tr |
− |
|
3 |
|
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
|
8a |
|
|
3b |
|
3 |
= 0.375 |
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
V − b |
|
V 2 |
|
|
|
|
|
|
|
|
− 1 |
|
|
|
|
|
|
|
|
27b 2 |
|
|
|
|
|
27bR |
|
|
8 |
|
|
||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3V |
|
V 2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
r |
|
|
|
|
|
|
r |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Дитеричи (1898) |
|
|
|
|
RT |
|
|
|
|
|
|
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
|
Tr e |
2− 2/T V |
|
|
|
|
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
a |
|
|
|
2 |
|
|
|
||||||||||||||||||
|
|
|
|
p = |
|
|
|
|
|
|
|
|
exp − |
|
|
|
|
|
|
|
|
|
|
pr |
|
= |
|
|
|
|
|
|
r r |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2b |
|
|
|
|
= 0.271 |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
V − b |
|
RTV |
|
|
|
|
|
|
|
2Vr |
− 1 |
|
|
|
|
|
|
4e |
2 |
b |
2 |
|
|
|
|
|
|
4Rb |
|
|
|
e |
2 |
|
|||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
|
Бертло (1900) |
|
|
|
|
|
|
|
|
RT |
|
|
|
|
|
|
a |
|
|
|
|
|
|
|
|
|
|
|
|
8Tr |
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
2 |
|
3 |
|
|
|||||||||||||
|
|
|
|
|
|
p = |
|
|
|
|
|
|
|
|
|
− |
|
|
|
|
|
|
|
|
|
|
p |
|
= |
|
|
|
|
|
|
− |
|
|
|
|
|
|
|
1 |
2aR |
1 |
2 |
|
2 a |
|
1 |
|
|
|
|
|
= 0.375 |
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3b |
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
V − b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
− 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
TV 2 |
|
|
|
|
r |
|
|
3V |
|
T V |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
|
|
|
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3b |
3 |
|
3 |
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
r |
|
|
|
|
|
r r |
|
|
12 |
|
|
|
|
3bR |
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
p = ( |
|
|
|
|
|
) |
|
|
|
|
|
|
( |
|
|
|
|
|
) |
|
, |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
Битти-Бриджмена (1927) |
|
|
1− γ |
|
RT |
|
|
|
V + β − α |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V 2 |
|
a |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
где α = a |
|
|
|
1 |
+ |
, |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
β = b |
|
|
1 |
− |
b |
|
, |
|
γ= |
|
|
|
c0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
V |
|
VT |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
Вириальное (1901) |
|
RT |
|
|
|
B2 |
|
|
|
|
B3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
(Камерлинг-Оннес) |
p = |
|
|
|
|
1+ |
|
|
|
|
|
|
+ |
|
|
|
+ |
... |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
V |
|
|
V |
|
V |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
термодинамики химической Основы .1 а в а л Г
19
20 |
Г л а в а 1. Основы химической термодинамики |
(1.5.а)
(1.5.б)
(1.6)
Наиболее известным уравнением, описывающим одновременно свойства и газа, и жидкости, является уравнение Ван-дер-Ваальса:
|
|
p = |
|
|
RT |
|
|
− |
a |
|
|
|
|
|
|
Vm − b |
|
|
|
||||||||
|
|
|
|
Vm2 |
|
|
|||||||
или |
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
RT |
|
2 |
a |
ab |
|
||||||
Vm |
− b + |
|
Vm + |
|
Vm − |
|
= 0 |
||||||
p |
|
p |
|||||||||||
|
|
|
|
|
p |
|
|||||||
Уравнение (1.5) – кубическое и, следовательно, имеет или три действительных корня, или один действительный и два мнимых. При температурах выше критической уравнение (1.5) имеет один действительный корень, и по мере повышения температуры кривые, вычисленные по уравнению Ван-дер-Ваальса, приближаются к гиперболам, соответствующим уравнению состояния идеального газа (рис. 1.3.б). При температурах ниже критической расчет по уравнению Ван-дер-Ваальса дает волнообразные кривые с тремя действительными корнями, из которых только два (точки 1 и 2) физически осуществимы. Третий корень (точка 3) физически нереален, поскольку находится на участке
кривой, соответствующем условию |
|
∂p |
> 0 . Согласно построению |
|
|
|
|
||
|
||||
|
|
∂V T |
|
|
Максвелла для того, чтобы расчетная кривая соответствовала экспериментальной равновесной изотерме, нужно вместо волнообразной кривой провести горизонтальную прямую так, чтобы отсекаемые ею площади под кривой были равны.
В критической точке изотермы реальных газов имеют точку перегиба, координаты которой можно определить из условий
|
∂p |
|
= 0, |
|
|
|
|
|
|
∂V |
|
|||
|
T =Tc |
|
||
|
|
|
|
|
|
∂ 2 p |
|
|
|
|
= 0. |
|||
|
|
|
|
|
|
2 |
|||
|
∂V |
|
|
|
|
|
T =T |
c |
|
|
|
|
|
|
Эти уравнения позволяют связать индивидуальные постоянные уравнения с критическими параметрами газа (см. пример 1-3 и таблицу П-1 в Приложении).
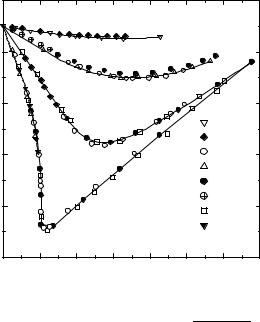
Вводя безразмерные приведенные переменные: давление pr = p/pc, объем Vr = V/Vc и температуру Tr = T/Tc, можно получить приведенное уравнение состояния реального газа, которое не содержит в явном виде индивидуальных постоянных (см. пример 1-3 и таблицу П-1).
Г л а в а 1. Основы химической термодинамики |
21 |
Состояния разных веществ, имеющие одинаковые значения приведенных переменных, называют соответственными. Согласно закону соответственных состояний, если для рассматриваемых газов значения двух приведенных переменных одинаковы, должны совпадать и значения третьей приведенной переменной. Таким образом, уравнения состояния различных веществ, записанные в приведенных переменных, должны совпадать. Это утверждение эквивалентно постулату о существовании общего универсального приведенного уравнения состояния:
|
|
|
|
|
|
|
|
F(pr, Vr, Tr) = 0. |
|
|
|
|
|
(1.7) |
|
||
|
Поскольку |
это |
уравнение |
|
1.1 |
|
|
|
|
|
|
|
|||||
не |
содержит |
в |
явном |
|
виде |
|
|
|
|
Tr |
= 2.0 |
|
|
|
|||
индивидуальных |
постоянных, |
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|||||||||
оно должно быть применимо к |
|
0.9 |
|
|
Tr |
= 1.5 |
|
|
|
||||||||
любому газу. Закон соответст- |
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
венных |
состояний |
является |
pV/RT |
|
|
|
|
|
|
|
|
||||||
общим утверждением, не свя- |
0.7 |
|
|
|
|
|
|
|
|||||||||
занным с |
конкретным |
видом |
|
|
|
T = 1.2 |
|
CH4 |
|
||||||||
уравнения состояния. На прак- |
|
|
|
r |
|
|
N2 |
|
|||||||||
Z = |
|
|
|
|
|
|
|
||||||||||
тике |
закон |
соответственных |
0.5 |
|
|
|
|
|
C2H4 |
|
|||||||
состояний |
приближенно |
вы- |
|
|
|
|
|
|
|
C2H6 |
|
||||||
|
|
|
Tr = 1.0 |
|
|
|
C3H8 |
|
|||||||||
полняется |
для |
однотипных |
|
|
|
|
|
|
|
||||||||
веществ, что позволяет, напри- |
|
0.3 |
|
|
|
|
|
H2O |
|
||||||||
|
|
|
|
|
|
C4H10 |
|
||||||||||
мер, использовать для реаль- |
|
|
|
|
|
|
|
CO2 |
|
||||||||
ных газов обобщенные диа- |
|
0.1 |
|
|
|
|
|
|
|
||||||||
граммы сжимаемости (рис. 1.4). |
|
1.0 |
2.0 |
3.0 |
4.0 |
5.0 |
6.0 |
7.0 |
|||||||||
|
|
|
|
|
|
|
|
|
|
0.0 |
|||||||
|
С высокой точностью по- |
|
|
|
|
|
pr |
|
|
|
|||||||
ведение любого реального газа |
Зависимость фактора сжимаемости |
|
Рис. 1.4 |
|
|||||||||||||
можно описать с помощью ви- |
|
|
|||||||||||||||
риального |
уравнения состоя- |
|
некоторых газов от приведенного |
|
|
|
|||||||||||
|
давления при разных приведенных |
|
|
|
|||||||||||||
ния – бесконечного ряда по |
|
|
|
|
|||||||||||||
|
|
температурах |
|
|
|
|
|||||||||||
степеням обратного объема: |
|
|
|
|
|
|
|
|
|
||||||||
|
|
RT |
|
|
B |
2 |
|
|
|
B |
3 |
|
|
|
||
p = |
|
|
1 |
+ |
|
|
|
+ |
|
|
+... |
(1.8.а) |
||||
|
|
|
|
|
|
|
|
2 |
||||||||
|
|
|
|
|
|
Vm |
|
|
|
|
|
|||||
|
|
Vm |
|
Vm |
|
|
|
|||||||||
или давления |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
RT |
|
|
|
' |
|
|
|
' |
|
2 |
|
|
|||
p = |
|
(1+ B2 p |
+ B3 p |
|
+...), |
(1.8.б) |
||||||||||
Vm |
|
|||||||||||||||
где Bi, Bi' – i-ые вириальные коэффициенты, которые зависят от природы газа и от температуры. Первый вириальный коэффициент равен 1. Так как в большинстве случаев B2/V >> B3 /V 2 >> ..., при описании экспериментальных данных ограничиваются вторым вириальным коэффициентом. При низких температурах B2 < 0, с ростом температуры его

22 |
|
|
|
Г л а в а 1. Основы химической термодинамики |
|
|
|
|
|||||||
|
|
значение проходит через ноль, затем становится положительным, дос- |
|||||||||||||
|
|
тигает максимума и далее очень медленно убывает. При высоких тем- |
|||||||||||||
|
|
пературах B2 и все последующие вириальные коэффициенты стремятся |
|||||||||||||
|
|
к нулю (рис. 1.5). Температура, при которой B2 = 0, называется темпе- |
|||||||||||||
|
|
ратурой Бойля. При этой температуре фактор сжимаемости реального |
|||||||||||||
|
|
газа близок к 1 при любых давлениях. Значения вторых вириальных ко- |
|||||||||||||
|
|
эффициентов некоторых газов при разных температурах приведены в |
|||||||||||||
|
|
приложении (табл. П-3). |
|
|
|
Термическое уравнение со- |
|||||||||
|
50 |
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
стояния |
можно |
|
определить |
||||
|
He |
|
|
|
|
|
|
|
экспериментально, |
изучая, на- |
|||||
|
|
|
|
|
|
|
|
пример, |
зависимость |
объема |
|||||
|
0 |
|
|
|
|
|
|
|
|||||||
|
|
|
CH4 |
Xe |
|
|
|
фазы от температуры и давле- |
|||||||
|
H2 |
Ar |
|
H2O |
|
ния. При этом получают вели- |
|||||||||
|
|
|
|
|
|
|
|||||||||
−1 |
|
|
|
NH3 |
|
|
чины, |
называемые |
|
термиче- |
|||||
-50 |
|
|
|
|
|
|
|
||||||||
3 моль |
|
|
|
|
|
|
|
скими коэффициентами: |
|
||||||
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
C H |
|
|
|
|
(1.9.а) |
|
∂V |
|
|
= |
−βV0 , |
|
, см |
|
|
|
3 8 |
|
|
|
|
|
|
|
||||
-100 |
|
|
|
|
|
|
|
|
|
∂p |
|
T |
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
B |
|
|
|
|
|
|
|
|
где β – изотермический коэф- |
||||||
|
|
|
|
|
|
|
|
|
|||||||
|
-150 |
|
|
|
|
|
|
|
фициент сжимаемости, |
|
|
||||
|
-2000 |
C2H6 |
|
|
|
|
|
(1.9.б) |
|
(∂∂VT )p |
= αV0 , |
||||
|
200 |
400 |
600 |
800 |
1000 |
1200 |
где α – изобарный коэффици- |
||||||||
|
|
|
|
T, K |
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
ент расширения, V0 – объем |
|||||||
|
|
|
Зависимость второго вириального |
|
|||||||||||
|
Рис. 1.5 |
|
|
при T = 0 K. |
|
|
|
|
|
||||||
|
|
|
коэффициента некоторых газов |
|
К термическим коэффици- |
||||||||||
|
|
|
|
от температуры |
|
|
ентам относится также частная |
||||||||
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
производная |
|
|
|
|
|
|
|
(1.9.в) |
|
|
|
|
∂p |
= γp , |
|
|
|
|
|
|
||
|
|
|
|
|
|
∂T |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
V |
|
|
|
|
|
|
|
|
|
|
где γ – изохорный коэффициент давления. |
|
|
|
|
|
|
|||||||
|
|
|
Последнюю величину для конденсированной фазы нельзя измерить |
||||||||||||
|
|
экспериментально, так как невозможно нагреть тело, чтобы не изме- |
|||||||||||||
|
|
нился его объем или объем оболочки, в которую оно помещено. Коэф- |
|||||||||||||
|
|
фициент γ можно рассчитать, используя цепочечное соотношение Эй- |
|||||||||||||
|
|
лера (см. Приложение IV): |
|
|
|
|
|
|
|
|
|
||||
(1.10)
откуда
(1.11)
|
∂p |
|
∂V |
|
∂T |
= −1, |
|
|
|
|
|
|
|
|
|
|
|||||||
|
∂V T |
∂T p |
∂p V |
|
|||
γ = βαp .

Г л а в а 1. Основы химической термодинамики |
23 |
Если известны термические коэффициенты, то уравнение состояния конденсированной фазы можно получить, интегрируя дифференциальное уравнение
|
∂V |
|
∂V |
dT . |
(1.12) |
dV = |
|
dp + |
|
||
|
∂p T |
|
∂T p |
|
|
Уравнение состояния идеального газа и вириальное уравнение состояния реального газа можно вывести методами статистической термодинамики (см. § 16).
ПРИМЕРЫ
Пример 1-1. Докажите, что при больших объемах уравнение Ван- дер-Ваальса переходит в уравнение идеального газа.
Решение. Уравнение Ван-дер-Ваальса для 1 моля:
p = |
RT |
− |
a |
. |
V − b |
|
|||
|
V 2 |
|||
При больших объемах вторым слагаемым в правой части можно пренебречь: a/V 2 → 0. В знаменателе первого слагаемого можно пренебречь постоянной b: V – b → V. В пределе получаем уравнение состояния идеального газа:
V →∞ |
RT |
|
p → |
|
. |
V |
||
Пример 1-2. Найдите вириальные коэффициенты Bi для газа, подчиняющегося уравнению Ван-дер-Ваальса.
Решение. В уравнении Ван-дер-Ваальса выделим сомножитель RT/V:
|
RT |
|
a |
|
RT |
|
|
|
1 |
|
|
a |
|
p = |
− |
= |
|
|
|
|
− |
. |
|||||
|
|
|
|
|
|
|
|
||||||
|
V − b |
|
V 2 |
|
V |
|
1 |
− |
b |
|
RTV |
||
|
|
|
|
|
|
|
|
|
|
V |
|
|
|
Если разложить первое слагаемое в скобке в ряд по степеням b/V, получим:
|
RT |
|
b n |
|
a |
|
|||
p = |
|
|
|
|
|
|
− |
|
. |
|
∑ |
|
|||||||
|
V |
|
|
|
|
||||
|
n=0 |
V |
|
RTV |
|||||
Из этого разложения следует, что второй вириальный коэффициент газа Ван-дер-Ваальса зависит от температуры:
B2 = b − RTa ,
а остальные постоянны: Bn =bn−1 .