

патфізіологія
.pdfГлава 3. Роль спадковості в патологічній фізіології
Клініко!генеалогічний метод. Сутність методу зводиться до виявG лення родинних зв’язків та простежування хвороби або ознаки у сім’ї. ГенеалогічнийметодналежитьдонайбільшуніверсальнихметодівгеG нетики людини. Технічно він складається з двох етапів: проведення родинного та генеалогічного аналізу. Складання родинних зв’язків починають від пробанда, яким називають особу, що першою потраG пила у поле зору лікаря. Зазвичай родовід складають за однією або кількома ознаками.
Метоюгенеалогічногоаналізуєвстановленняспадковогохарактеру ознакитатипуспадкування.Наприклад,родовідродиниРомановихдозG воливпростежити,звідкивцарськійродиніз’явиласягемофілія,наяку хворівспадкоємецьМиколиІІОлексій:початокїйпоклалаанглійська королева Вікторія. Гемофільний ген виник у неї, напевно, в результаті мутації. Через те, що королева була гетерозиготною, вона була цілком здороваіпрожиладопохилоговіку.ОднакунеїбувсинGгемофілітикта двідоньки—носіїцьогогена.Видавшидочокзаміж,Вікторія«ощаслиG вила»двіцарськіродини—РосіїтаІспанії.ЖінкаМиколиІІбулаонуG коюВікторії.Вонаіпередалагемофіліюспадкоємцюпрестолу.
В результаті аналізу можна з’ясувати, є ознака домінантною чи реG цесивною; чи зчеплена вона зі статтю; чи є різниця успадкування озG наки жінками та чоловіками та ін.
Об’єктивне встановлення типу успадкування того чи іншого заG хворювання можливе лише за умови масового обстеження сім’ї.
Івцьомувідношеннігенеалогічнийметоднадаєнеоціненнудопомогу. Близнюковий метод дозволяє судити про відносну роль спадкоG
востійсередовищавмінливостірізнихознакорганізму.МетодбазуєтьG ся на порівнянні ознак, що зустрічаються у різних груп близнюків. При цьому проводять порівняння монозиготних близнюків із дизиG готними, партнерів монозиготних пар між собою, беруть результати аналізу близнюкової виборки і загальної популяції.
Монозиготними (однояйцевими, ідентичними) близнюками наG зиваютьіндивідів,щовирослизоднієїзиготи,якаподілиласянаранніх стадіях дроблення на дві частини. Дизиготні (двояйцеві, неідентичні) близнюкивиникаютьунаслідокзаплідненнядвоходночаснодозрілих яйцеклітин. БудьGяка якісна ознака може зустрітися або у обох близG нюків даної пари, або у одного з них.
Впершомувипадкупаразветьсяконкордантною,вдругому—дисG кордантною.СтупіньконкордантностізаспадковообумовленимиознаG ками буде вищою у ідентичних близнюків. Порівняння ступеня конG кордантностіудвохгрупблизнюківдозволяєробитивисновкипровідносG нийхарактерспадковостійсередовищавконкретнійформіпатології.
61

Розділ І. Нозологія
Популяційно!статистичний метод полягає в дослідженні ознак у веG ликих групах, що розрізняються за спадковими характеристиками (раси, національності, етнічні групи та ін.) або за умовами життя. ПоG пуляційноGстатистичний метод дозволяє вивчити значення спадкоG вих хвороб, роль спадкового середовища у розвитку хвороб зі спадкоG вими схильностями, причини різної частоти спадкових хвороб у геоG
графічних зонах та різних популяціях.
Цитогенетичні методи мають вагоме значення у вивченні спадкоG воїпатології.Донихналежать:аналізкаріотипу(виконуєтьсяпризнаG ходженні у пробанда в ході клінічного обстеження ознак будьGяких хромосомних хвороб, у дітей з багатьма вродженими вадами незрозуG мілої етіології, у подружжя при багаторазових спонтанних абортах,
умертвонароджених, при визначенні прогнозу здоров’я потомства,
увипадках, коли в роду є люди з хромосомними хворобами), аналіз статевогохроматинутільцяБера(застосовуєтьсяпривиявленніупроG банда ознак будьGяких хромосомних хвороб у ході клінічного обстеG ження,ужінокзпорушеннямрепродуктивної(дітородної)функціїнеG зрозумізого генезу та ін.).
Порядзпереліченимисутогенетичнимиметодами,взалежностівід встановленогозавдання,прививченніспадковоїпатологіївсебільшого поширеннянабуваютьрізніпрактичніметодидослідження,щодозволяG ютьосягнутисутністьпатогенезухвороби.Донихналежатьбіохімічні, імунологічні,цитологічніметоди,методвивченнярельєфнихвузлівна
шкірі,атакожекспериментальнемоделюванняспадковиххвороб. Біохімічні методи застосовують в основному в тих випадках, коли
мають підозру на спадкові хвороби обміну речовин, на ті форми спадG кових хвороб, при яких визначені вади первинного генного продукту
або патогенетична ділянка розвитку захворювання.
Імунологічні методи реальні для діагностики поки що невеликої групи спадкових хвороб (глікогенози, гемоглобінопатії, іхтіоз та ін.), хоча можливості їх достатньо великі. Клітини хворих можна вивчати безпосередньо або після культування цитохімічними, імунофлуоресG
центними, електронноGмікроскопічними методами.
Метод вивчення рельєфних вузлів на шкірі, що утворюються капіG лярними лініями та гребінцями, або дерматогліфіка (дерма — шкіра, гліфе—гравірувати),базується на індивідуальномухарактері капілярG
ного рисунка, який знаходиться під генетичним контролем.
Експериментальне моделювання спадкових хвороб базується на штучному розмноженні мутаційних ліній тварин, що мають ті чи інші спадкові дефекти, аналогічні таким вадам у людини (ахондроплазія у кроликів, гідроцефалія та дефекти губи у мишей, гемофілія у собак).
62

Глава 3. Роль спадковості в патологічній фізіології
3.5. Характеристика спадкових хвороб
Розрізняють хвороби: генні, хромосомні, зі спадковими схильноG стями.
Для спадкових хвороб характерний клінічний поліморфізм. Він виявляється в різних строках прояву симптомів або початку захворюG вання, різному ступені виявлення хворобливих ознак, неоднакових термінах летального кінця. Варіації прояву спадкових хвороб не обG межуються тільки клінічними характеристиками. Вони виявляються також у коливаннях біохімічних та інших показників, які входять до загальногопоняттяфенотипу.Так,захворюваннязнедосконалимостеоG генезом характеризується ламкістю кісток, прогресуючим отосклероG зом та блакитними склерами, але ці три головні ознаки зустрічаються лише у 44 % носіїв гена, ламкість кісток — у 63 %, глухота — у 60 %.
Таким чином, у різних хворих основні патологічні симптоми комG бінуються поGрізному. Значно може коливатися і час прояву хвороби. Так,середнійвікпочаткупроявузахворюваннянахореюГетінгтона— 40–45 років, але у деяких хворих вона з’являється уже в перші роки життя, а інколи й після 60 років.
3.5.1. Генні хвороби (молекулярно!генетичні)
Оскільки генна мутація в порівнянні з хромосомною займає поG рівняно невелику ділянку генетичного матеріалу, то зазвичай супроG воджуєтьсяменшгрубимипорушеннями.РепродуктивнафункціяноG сія при цьому зберігається, і тому такі захворювання частіше передаG ються в поколіннях, тобто є спадковими в повній мірі.
За домінантним типом успадковуються різні скелетні та інші аноG малії,щонеперешкоджаютьрозмноженню,нескорочуютьтривалості життяітомумалопіддаютьсявідбору.Такимианомаліямиможутьбути короткопалість,багатопалість,пальці,щовикривленіабозрослися,виG кривленнянігтів,відсутністьбоковихрізців,короткозорість,астигматизм.
За домінантним типом передаються такі тяжкі хвороби, як вродG жена катаракта, отосклероз, деякі форми м’язової атрофії, прогресуG юча хорея Гетінгтона, ахондроплазія, що характеризується карликоG вим ростом та непропорційною будовою тіла.
ДонайбільшнебезпечниххворобцієїгрупиможнавіднестичисленG нийполіпозтовстогокишечника(якиймаєтенденціюдопереродження взлоякіснупухлину)танейрофіброматоз(хворобаРеклінгаузена).
Більшість спадкових хвороб передаються за рецесивним типом. Хворобапроявляєтьсятоді,колидітиодержуютьпатологічнийгенобох батьків. Самі ж батьки, що є гетерозиготними носіями ознаки, залиG шаються фенотипічно здоровими. Велике значення для прояву цих
63

Розділ І. Нозологія
хвороб у спадкоємців має кровна спорідненність батьків, у яких є веG лика ймовірність володіти однаковим рецесивним патологічним геG ном. До таких хвороб належать дефекти амінокислотного обміну (феG нілкетонурія, альбінізм, алкаптонурія), вроджена глухонімота, мікроG цефалія, пігментопатія (дефіцит ферменту та блок на певному етапі метаболізму). Так, підвищена чутливість до алкоголю деколи пов’язаG на з низькою активністю ферменту алкогольдегідрогенази. Дефекти ферментів лікарського метаболізму призводять до підвищеної чутлиG
вості до лікарських препаратів і т. ін. Відомі такі типи спадковості:
—аутосомноGдомінантний — фенотипічно патологічний стан — виявляється у гетерозигот;
—аутосомноGрецесивний—фенотипічнопатологічнийстан—виG являється у гомозиготному стані;
—ХGзчеплене спадкування — генилокалізовані в ХGхромосомі (ХGзчеплений тип спадкування, ХGзчеплений домінантний тип спадG кування).
Етіологічним фактором генних хвороб є генні мутації. У людини приблизно 50–100 тис. генів, і кожен ген може мутувати та зумовлюG вати іншу структуру білка. Отже, кількість спадкових хвороб генної природи може бути дуже великою. Кожна генна мутація зумовлює зміну або відсутність білка. Наприклад, при галактоземії різко знижеG наактивністьферментугалактозоG1GфосфатGуридилтрансферази,внасG лідок чого в клітинах накопичується галактозоG1Gфосфат. Ця сполука пригнічує ферментативні реакції вуглеводного обміну за участю фосG форильованихпроміжнихпродуктів.ПісляцьоговідбуваєтьсяураженG няпечінки,мозку,загальнепорушеннярозвитку.Катаракти,характерні для цього захворювання, утворюються через велику концентрацію гаG лактозоG1Gфосфатуврідинахорганізмутаутвореннягалактитолу.
Для багатьох генних хвороб уже ідентифіковано первинний аноG мальний продукт гена — ключова патогенетична ділянка на біохімічG ному рівні.
Виходячи з цього, хвороби класифікуються за біохімічним потенG ціалом, у залежності від того, які білки уражені — структурні, трансG портні чи ферментні.
Прикладом захворювання, при якому зміни стосуються структурG них білків, може бути синдром Елерса — Данлоса, при якому порушеG на структура колагену. Синдром характеризується підвищеною еласG тичністю шкіри, збільшенням рухомості суглобів, підвищеним розтяG гуванням хорд серцевих клапанів, відшаруванням сітківки, підвивиG хом кришталика.
64

Глава 3. Роль спадковості в патологічній фізіології
УраженнятранспортнихбілківвідзначаєтьсяприлізинуричнійнеG переносимості білка, при якій порушується транспорт діамінокислот (лізину, аргініну, орнітину) в ниркових канальцях.
Найбільш поширеною та достатньо вивченою групою моногенG нихзахворюваньєензимопатії.Озброєннягіпотезою«одинген—один фермент» призвело до розшифровки багатьох з них. Ензимопатії, при яких розшифровано первинний дефект ферменту, поділені на хвороG би накопичення ліпідів, глікогену, глікопротеїнів, порушення аміноG кислотного,вуглеводного,пуриновоготапіримідиновогообмінів,поG рушення гормоногенезу та ін.
Загальна частота генних хвороб в популяціях в цілому дорівG нює 1–2. Частота окремих форм коливається від 1:2000–3000 (мукоG вісцидоз) до 1:100 000 (атаксія — телеангіоектазія). Умовно частоту генноїхворобиможнавважативисокою,якщозустрічаєтьсяодинхвоG рий на 10 000 новонароджених, а низькою — 1:40 000. Так, за рівнем спадкових хвороб обміну речовин (фенілкетонурія, тирозинемія, гаG лактоземія та ін.) частота належать до категорії середніх і низьких.
3.5.2. Хромосомні хвороби
Більшістьхромосомнихабераційдаєнежиттєздатну статевуклітиG ну. Однак, якщо життєздатність її зберігається і вона підлягає заплідG ненню, плід може загинути на різних стадіях свого розвитку. Разом з тим, якщо хромосомний дисбаланс сумісний з постнатальним існуG ванням, то звичайно порушено соматичний та психічний розвиток організму. В тих рідких випадках, коли здатність відтворення потомG ства збережено (3–5 %), нащадки успадковують цю ж патологію.
Хромосомні хвороби можна розподілити на дві великі групи: виG кликані геномними мутаціями, тобто змінами кількості хромосом (поліплодії, анеуплодії) при збереженні структури останніх, та хвороG би,зумовленіхромосомнимимутаціями,тобтозмінамиструктурихроG мосоми(транслокації,інверсії,делеції).ХромосомнахворобаможевиG никнути внаслідок мутацій у клітинах ембріона на ранніх стадіях його розвитку (особливо на стадіях дроблення зиготи).
У людини розкриті всі форми геномних та хромосомних мутацій. ПовніформитетраGтаполіплодійвиявленітількиприспонтаннихаборG тах,щосвідчитьпроїхлетальнийефектнаранніхстадіяхрозвитку.ЗаG гальнакількістьописанихтипівхромосомниханомалійулюдинитільки гаметичного походження — біля 750, з яких понад 700 — структурні перебудови.Достуктурнихперебудовзалучаютьсявсіхромосоми.
Багатогранністьописанихформхромосомниханомалійлюдини— добре встановлений факт. Однак не всі вони можуть розглядатися як самостійні клінічні синдроми.
65

Розділ І. Нозологія
Сьогодні виділяють такі групи та види хромосомних синдромів, щодобрерозпізнаються:синдромимоносомій,ХGмоносоміяабосинG дром Шерешевського — Тернера, синдроми трисомій 8+, 9+, 13+ (синG дром Патау),18+ (синдром Едвардса), 21+ (синдром Дауна), полісомії застатевимихромосомами:ХХХ(ХGтрисомія),XXV(синдромКлайнG фельтера).Немовлята,трисомнізахромосомою13+,маютьхарактерні аномалії, в тому числі анофтальм (мікрофтальм), вовчу пащу, заячу губу, вроджені вади серця.
Існує велика кількість синдромів, зумовлених діленням (наприкG лад, синдром «котячого крику»). Ці синдроми мають чітку клінічну картину. Так, характерна форма голови й обличчя у хворих, клінодакG тилія, м’язова гіпотонія в поєднанні з розхитаністю суглобів є в комG плексі у всіх хворих з синдромом Дауна, хоч кожна ознака поодинці можебутивідсутняв20–30%випадків.РозумовавідсталістьспостеріG гається звичайно в ступені імбецільності. Близько половини хворих мають пороки серця та великих судин. При хворобі Дауна змінений стан гуморального та клітинного імунітету, що зумовлює підвищену чутливість хворих до інфекції. У 94 % хворих виявляється каріотип 47XX/ХV/21,тобтопростаформатрисоміїаутосоми21.Близько4–5% має транслокаційну форму трисомії. Загальною для всіх форм хромоG сомних хвороб є багатогранність уражень: черепноGмозкові дисгарG монії, вроджені вади розвитку внутрішніх та зовнішніх органів, упоG вільнений ріст та розвиток, відставання у психічному розвитку, поруG шення функцій нервової та ендокринної системи.
3.5.3. Хвороби зі спадковою схильністю
Хвороби зі спадковими схильностями відрізняються від спадкоG виххворобтим,щодлясвогопроявленняпотребуютьдіїфакторівзовG нішнього середовища. Вони являють собою групу, багатогранну за нозологічними формами. За генетичною природою це дві групи хвоG
роб: моногенні та полігенні хвороби зі спадковими схильностями.
Моногенні хвороби зі спадковими схильностями — це схильності, пов’язані зі спадковою мутацією одного гена. Для свого прояву схильність обов’язково потребує обов’язкової дії фактора зовнішньоG го середовища, що зазвичай ідентифікується і відносно цієї хвороби може розглядатися як специфічний.
Моногенні форми спадкової схильності проявляються при дії різнихфакторівсередовища:забрудненняповітря,харчовихпродуктів, ліків. Відомо, наприклад, що деякі харчові продукти можуть викликаG ти патологічний стан у людей зі спадковою схильністю. Так, у деяких людей після вживання молока або молокопродуктів спостерігається
66

Глава 3. Роль спадковості в патологічній фізіології
утвореннягазів укишечнику. Впопуляції європеоїдної раситакііндиG віди складають 10–20 %, а у східних народів, негрів та індіанців АмеG рики — 70–100 %. Це пов’язано з аутосомною рецесивною ознакою несприймання лактози. В кишечнику дорослих гомозигот відсутня бетаGгалактозидаза, тому молочний цукор не засвоюється, а під дією
мікрофлори кишечника підлягає бродінню.
Полігенні хвороби зі спадковими схильностями визначаються багатьG ма генами, кожний з яких є скоріше нормальним, ніж патологічним. Визначення цих генів дуже ускладнене: своє патологічне виявлення вони здійснюють у взаємодії з комплексом факторів зовнішнього сеG редовища.
Не зважаючи на те, що полігенні хвороби зі спадковими схильноG стями є найбільш поширеною групою (гіпертонічна хвороба, атероG склероз, виразка шлунка, шизофренія, бронхіальна астма, псоріаз та ін.), механізм їх виникнення мало вивчений.
3.6. Важливість спадкових факторів у патології людини
Спадкові хвороби зустрічаються практично у всіх медичних галуG зях. Це численні хвороби внутрішніх органів, обміну речовин, систеG ми крові, ендокринної системи, печінки, очей, сечостатевої системи, нервові та психічні захворювання й тощо. Наприклад, у дерматології відомо майже 250 спадкових захворювань, в офтальмології та клініці нервових захворювань — до 200.
Дуже суттєвим є поширення спадкових хвороб у популяції людиG ни. Так, лише генні хвороби складають 1,5–2 %. Ще більш виражеG ною стає картина при аналізі контингенту хворих у клініках, особлиG во педіатричних. Встановлено, що в дитячих клініках 25–30 % дітей — це хворі зі спадковими патологіями. Зараз із хромосомними хворобаG ми народжуються 0,7 % усіх живонароджених немовлят. Основна частина ембріонів із хромосомними аномаліями гине ще до свого наG родження, обумовлюючи понад 400 спонтанних абортів і близько 6 % мертвонароджених.
Існують широко розповсюджені хвороби зі спадковими схильноG стями (атеросклероз, гіпертонічна хвороба, цукровий діабет, шизоG френія та ін.). Так, згідно з даними Міжнародної федерації з діабету,
в світі нараховуються десятки мільйонів хворих на цукровий діабет.
Принципи лікування і профілактики спадкових хвороб та хвороб зі спадковими схильностями. Визнання ролі факторів зовнішнього сеG
редовища в проявах спадкової патології дозволяє розробляти різні підходи до лікування хворих зі спадковими хворобами. Як і при лікуG ванні будьGякої хвороби, можна виділити три основні підходи до лікуG вання хворих зі спадковими хворобами та спадковими схильностями.
67

Розділ І. Нозологія
1. Симптоматичне лікування — лікарське, хірургічне (видалення або заміна уражених ділянок, наприклад, поліпозу прямої кишки, коG
рекція при незростанні верхньої губи, при вроджених пороках серця). 2. Патогенетичне лікування — корекція обміну речовин (признаG
чення дієти з низьким вмістом фенілаланіну при фенілкетонурії запоG бігаєрозвиткурозумовоївідсталості);звільненнявідпродуктівобміну, які є субстратом патологічної реакції (виведення заліза при таласемії, гемосорбціяприродиннійгіперхолестеринемії);хірургічнешунтуванG ня (накладення анастомозу між воротною та нижньою порожнистою веною при глікотеріозі, введення антигемофільного глобуліну при геG
мофілії, інсуліну при цукровому діабеті) і т. ін.
3. Етіологічне лікування. Завдяки успіхам молекулярної біології та генноїінженерії,проньогоможнаговоритиякпроперспективу:можG ливим буде включення штучно отриманих генів до геному людини; використанняспрямованогохімічногомутагенезу,щодозволитьіндуG куватиспецифічнімутаціївчітковизначеномулокусі(одержаннязвоG ротних мутацій — від патологічного алеля до нормального); виправG лення генетичного дефекту трансформацій у еукаріотів.
Профілактика спадкової патології в цілому, безумовно, є важлиG вим завданням сучасної медицини та організації охорони здоров’я. Взагальнійформіпрофілактикаповиннабутиспрямовананатритипи спадкової патології:
а) спадкові хвороби, що заново виникають як результат спонтанG ної мутації в зародкових клітинах батьків;
б) хвороби, що успадковані від попередніх поколінь; в) хвороби, що розвиваються внаслідок спадкової схильності та
дії шкідливих факторів зовнішнього середовища.
Серйознапроблемапрофілактики—індукованиймутагенезчерез дію хімічних, фізичних та біологічних факторів середовища, кількість яких постійно збільшується у зв’язку з науковоGтехнічним прогресом. Оскільки індукований мутагенез призводить до підвищення частоти спадкових хвороб, то він, з точки зору профілактичної медицини, поG винен бути повністю виключений. На цей час розроблена система геG нетичних та гігієнічних заходів із виключенням мутагенних факторів.
Найбільш поширеним та ефективним підходом до профілактики спадкових хвороб є медикоGгенетичне консультування. Сутність його полягає у визначенні прогнозу народження дитини зі спадковою хвоG робою, у поясненні ймовірності такої патології та консультації родині щодо прийняття рішення про народження дитини.
Треба розрізняти два терміни, що нерідко використовуються як синонім терміну «спадкова хвороба»:
68

Глава 3. Роль спадковості в патологічній фізіології
1.Термін «уроджені хвороби», з одного боку, — ширше поняття, ніж «спадкові хвороби», бо вродженими є всі хвороби, з якими дитина з’являється на світ, у тому числі й індуковані шкідливими хвороботG ворнимифакторамипривнутрішньочеревномуперіоді(віруснітабакG теріальніінфекції,травми,вадирозвиткуекзогенноїприроди).ЗіншоG го боку, не всі спадкові хвороби є вродженими. Дуже багато хвороб (хореяГетінгтона,м’язовадистрофіятаін.)з’являютьсяубільшпізньоG му (деколи дорослому) віці.
2.Термін «родинні хвороби» також не замінює поняття спадкових хвороб, оскільки має більш широке значення, тому що охоплює
іспадкові, і неспадкові хвороби, що зустрічаються в декількох членів родини під впливом однакових для них шкідливих факторів зовнішG
нього середовища.
Хвороба Дауна (рис. 3.1). Вивчення каріотипу показало наявність трисоміїза21Gоюпароюхромосом.Загальнакількістьхромосом—47, хоч може бути й 46 (це означає, що зайва 21Gша хромосома злилася з однією з великих, наприклад, із 15Gою). При цьому спостерігається розумова відсталість; характерна зовнішність — низький зріст, коротG копалі руки й ноги, монголоїдний розріз очей; аномалії внутрішніх органів, особливо серця. Хвороба Дауна зустрічається досить часто — один випадок на 500–600 родів. Жінки з хворобою Дауна інколи маG ють дітей. Оскільки це домінантна ознака, 50 % дітей, що народилися від таких матерів, здорові, а 50 % хворіють на ту саму хворобу.
а |
б |
в |
Рис. 3.1. Діти з синдромом Дауна:
а — європеоїд; б — негр; в — представник азіатської раси. Спільні ознаки синдрому Дауна більш помітні, ніж расові відмінності
(Фогель Ф., Могульский А. Генетика человека. — М.: Мир, 1989)
69
Розділ І. Нозологія
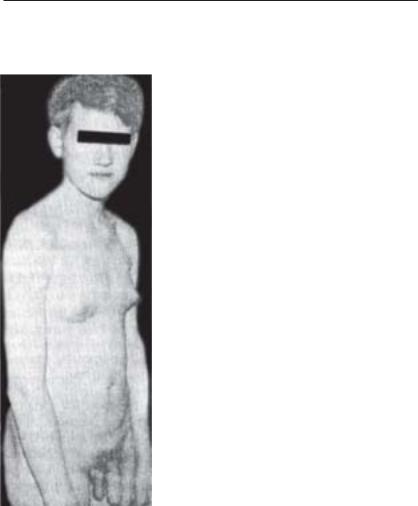
Синдром Клайнфельтера (рис. 3.2). Захворювання спостерігається учоловіків,йогочастота1:1000.Загальнакількістьхромосом47(каріоG тип ХХY), але трапляються і 48 (ХХХY), 49 (ХХХХY). Зовнішні стаG теві органи сформовані за чоловічим тиG пом. Характерним є високий зріст, астеніG чнабудоватіла,довгіноги,зниженнясперG матогенезу. Як і для інших хромосомних хвороб, властива розумова відсталість.
Усоматичних клітинахвиявляється статеG вийхроматин(тільцяБарра),кількістьякоG
годорівнюєкількостіХGхромосоммінус1.
Синдром Тернера (Шерешевського — Тернера) (рис. 3.3). Синдром розвивається
тоді, коли в жіночому організмі замість двохстатевиххромосом (ХХ)є тількиодна ХGхромосома.Загальнакількістьхромосом— 45(каріотип45,Х0).ЗовнішністатевіоргаG ни сформовані за жіночим типом. ХаракG терним є низький зріст, широка щитопоG дібна грудна клітка, недостатній фізичний і статевий розвиток. Внутрішні статеві органи недорозвинені, замість яєчників, наприклад, тільки фіброзні тяжі. У клітиG нах слизової оболонки рота немає статеG вогохроматину,щодопомагаєвстановити діагноз.
|
Трисомія за Х!хромосомою (каріо! |
|
|
тип ХХХ). У клітинах два тільця Барра. |
|
|
У хворих жінок спостерігається недорозG |
|
|
виненість яєчників, однак менструальний |
|
Рис. 3.2. Синдром |
цикл не завжди порушений; інколи жінки |
|
мають дітей. |
||
Клайнфельтера |
||
(за А.С. Єфимовим |
НосіїхромосомногонаборутипуY0гиG |
|
та співавт. К., 1983) |
нутьнаранніхстадіяхвнутрішньоутробноG |
горозвитку.ВивченняхромосомниханомалійдаєзмогудійтивисновG ку, що дефіцит аутосом небезпечніший, ніж їх надлишок, оскільки випадки моносомії поки що не описані.
Виявлення ролі спадковості в походженні захворювань має велиG ке значення для вибору методу лікування даного хворого, для прогноG зу потомства в нього і його батьків. Наприклад, у людини з брахідакG тилією (домінантна ознака) імовірність появи цього дефекту в дітей
70