

monomers-Платэ-книга
.pdf31
вательные бимолекулярные реакции отрыва водорода небольшими активными радикалами (Н , СН3 и в некоторой степени С2Н5 ) от молекул углеводородов и распада образующихся при этом макрорадикалов. Распад радикалов
происходит в основном по связи С—С в -положении к атому, имеющему неспаренный электрон. Энергетика распада также благоприятствует разрыву
С—С-связи; связи -С—Н в радикалах диссоциируют только в том случае,
если связи -С—С отсутствуют, например в С2Н5 или (СН3)3. Обрыв цепей осуществляется по реакциям рекомбинации или диспропорционирования ра-
дикалов в объеме реактора либо на его стенках. |
|
|
Пиролиз бутана протекает в частности по схеме |
|
|
C4H10 2 C2H5, |
C4H10 CH3 + н-C3H7, |
|
C2H5 C2H4 + H , |
|
|
C2H5 + C4H10 C2H6 + C4H9, |
CH3 + C4H10 CH4 + C4H9, |
|
H + C4H10 H2 + C4H9 |
н-C3H7˙ C2H4 + CH3 |
|
C4H9 C2H4 + C2H5 |
C4H9 C3H6 + CH3 |
|
H + C3H6 CH3 + C2H4 |
|
|
Состав продуктов пиролиза циклических углеводородов, например циклогексана, в целом аналогичен составу продуктов пиролиза алканов. В несколько больших концентрациях присутствуют водород, метан, этилен и пропилен и в значительных количествах — бутадиен-1,4.
Этилен подвергается термическим превращениям уже при сравнительно низких температурах, начиная примерно с 700 К. При низкотемпературном пиролизе этилена (800-1000 К) протекают реакции димеризации, полимеризации и самогидрирования. Основные продукты реакции – этан, пропилен, бутены. В меньших количествах образуются метан и бутадиен.
Среднетемпературный пиролиз (1000-1300 К) представляет наибольший интерес, так как именно в этом интервале температур на практике осуществляется производство этилена. Основными продуктами термических превращений этилена являются ацетилен и бутадиен. При высокотемпературном пиролизе (выше 1300 К) преобладающим направлением процесса становится дегидрирование этилена в ацетилен, причем с повышением температуры селективность образования ацетилена резко возрастает.
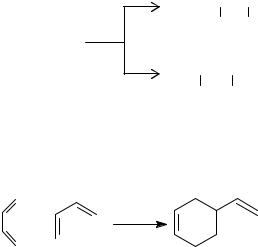
Аллен – простейший диеновый углеводород – образуется при пиролизе различного сырья в небольших количествах (<0,5%). Источником образования аллена является реакция распада аллильного радикала:
СН2=СНСН2  СН2=С=СН2 + Н.
СН2=С=СН2 + Н.
При относительно низких температурах (800 К) аллен вступает в реакции димеризации:
32
H2С—С=CH2
H2C=С—СH2
2CH2=C=CH2
H2С—С=CH2
H2С—C=СH2
Бутадиен-1,4 является целевым продуктом пиролиза различного углеводородного сырья. Основное направление термических превращений бута- диена–1,4 при температурах ниже 900 К – димеризация в 4- винилциклогексен, протекающая с высокой селективностью:
+
Термические превращения ацетилена начинаются уже при температурах ~ 600 К. Между тем весьма высокие энергии диссоциации связей С≡С и С—Н в ацетилене трудно согласовать со значительной реакционной способностью ацетилена при столь низких температурах. Принято считать, что механизм термических превращений ацетилена сильно отличается от механизма пиролиза алканов. При температурах ниже 1000 К основным направлением процесса является конденсация с образованием ароматических углеводородов: бензола, его производных и конденсированных углеводородов. С повышением температуры основными продуктами становятся высшие ацетиленовые углеводороды.
Продуктами глубокой конверсии алифатического сырья являются ароматические углеводороды. Термическая стабильность ароматических углеводородов значительно выше, чем термическая стабильность алифатических соединений и нафтенов. Ниже приведены эффективные константы скорости k (c-1) гомолитического распада С—С-связей для некоторых углеводородов при 1073 К:
н-гексан |
циклогексан |
бензол |
толуол |
18 |
1,5 |
0,001 |
0,003 |
Поэтому ароматические углеводороды, содержащиеся в подвергаемых пиролизу нефтяных фракциях, при 1000-1200 К и времени реакции ~ 0,1 с почти не претерпевают термического распада, затрагивающего ароматическое ядро. Они участвуют главным образом в реакциях конденсации и уплотнения.
Общие сведения о технологии процесса
Выход ненасыщенных газообразных углеводородов увеличивается при следующих условиях:
- кратковременном пребывании реагентов в горячей зоне (доли секунды) с последующим быстрым охлаждением (закалка) продуктов пиролиза;
33
- разбавлении сырья водяным паром для понижения парциального давления углеводородов (разбавление сырья паром используют для уменьшения скорости вторичных реакций и количества тяжелых жидких продуктов и кокса).
В настоящее время промышленный пиролиз проводится исключительно в трубчатых печах.
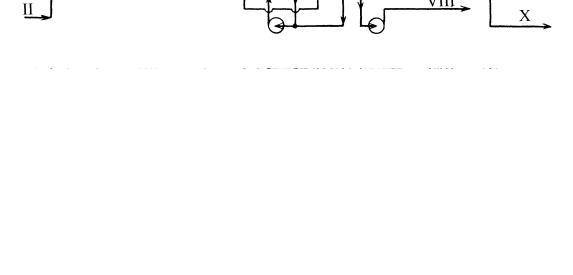
На рис. 1.1 показана принципиальная технологическая схема установки пиролиза.
Бензин, нагретый в теплообменнике и предварительно перемешанный с водяным паром, подается в трубчатую печь 1. Газ удаляют из печи при 1113-1123 К и подвергают быстрому охлаждению в закалочном аппарате 2, который представляет собой конденсатор смешения; в конденсатор подают водяной конденсат. При этом температура газа снижается на 423о, что оказывается достаточным для прекращения реакции пиролиза. Далее газ охлаждается до 673 К, и парожидкостная смесь разделяется в ректификационной колонне 3 на газ пиролиза и смолы.
Рис. 1.1. Принципиальная технологическая схема установки пиролиза бензина
1-печь; 2-закалочный аппарат; 3- ректификационная колонна. Потоки: I – водяной пар; II – бензин; III – конденсат; IV – вода; V – поглотительное масло; VIтяжелое масло; VII – газ на очистку; VIII – вода на очистку; IX – пар; X – легкое масло
Ниже приведены данные о выходе продуктов пиролиза (в %(мас.)), протекающего при 1113-1143 К и времени контакта 0,25-0,40 с:
Водородно-метановая фракция …………. 17,7
Этилен ……………………………………. 25,5 Пропилен …………………………………. 16,2 Пропан ……………………………………. 1,0 Бутан-бутены ………………………….. 12,2 Фракция н.к.-423 К ……………………… 19,9

34
Фракция 423-523 К ………………………. 1,5 Остаток выше 523 К ……………………… 3,3 Потери …………………………………….. 2,7
Бензины-рафинаты, выкипающие до 423 К, содержат 50-55% (мас.) изоалканов, 30-35% н-алканов, 4-8% циклоалканов, 2-4% ароматических углеводородов.
Другие методы пиролиза
Необходимость расширения сырьевой базы, сокращения расхода сырья, энергетических и материальных затрат привела к разработке новых модификаций процесса, рассчитанных в основном на пиролиз тяжелых углеводородов. Кроме того, сегодня разрабатываются новые процессы пиролиза со значительно более высоким выходом этилена: пиролиз в присутствии инициирующих добавок, гидропиролиз, высокотемпературный пиролиз с использованием газообразных теплоносителей (водяной пар, дымовой газ, водород), пиролиз в расплаве металлов и солей, пиролиз в присутствии гетерогенных катализаторов – каталитический пиролиз.
Гетерогенно-каталитический пиролиз проводят в мягких условиях (1053-1063 К) в присутствии в качестве катализатора ванадия на носителе, модифицированном добавками, снижающими коксообразование. При этом достигается более высокий выход этилена за один проход.
1.1.6. Коксование
Основная цель процесса коксования – получение светлых нефтепродуктов и кокса из гудрона и тяжелых нефтяных остатков. В зависимости от вида сырья и условий процесса можно получить топливный, анодный или игольчатый кокс. На рис. 1.2 показана принципиальная схема установки замедленного коксования фирмы "АББ Луммус Крест".
35
Рис. 1.2. Принципиальная технологическая схема установки замедленного коксования фирмы "АББ Луммус Крест"
1 - ректификационная колонна; 2- нагревательная печь; 3- камеры коксования; 4 - сепаратор; 5 - блок разделения бензина и газов.
Потоки: I – сырье; II – пар; III - топливный газ; IV– углеводороды С3-С4; V – бензин; VI - легкий газойль; VII – тяжелый газойль
Сырье после нагревания подают в ректификационную колонну 1 для извлечения легких нефтяных фракций, содержащихся в сырье, и извлечения тяжелых фракций – рециркулята – из паро-продуктовой смеси, поступоющей из камер коксования 3.Рециркулят смешивают с сырьем, затем он поступает из нижней части колонны в нагревательную печь 2, где сырьевая смесь нагревается до температуры 753-783 К. После печи она направляется в необогреваемые камеры 3. Газо-продуктовая смесь выходит из верхней части камер коксования и поступает в ректификационную колонну 1, где разделяется на газ, бензин, легкий и тяжелый газойли. Газ и бензин выходят сверху из колонны, проходят сепаратор 4 и в колонне 5 разделяются на топливный газ, пропан-пропиленовую, бутан-бутиленовую фракции, легкий и тяжелый бензин. Кокс из камеры 3 удаляется водой под высоким давлением.
Выбор технологии коксования определяется тем, какие именно нефтепродукты являются целевыми. Замедленное коксование проводится для увеличения выхода нефтяного кокса, коксование в кипящем слое – для увеличения выработки светлых продуктов.
1.2. КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Основными процессами углубленной переработки нефти являются каталитические процессы: гидрокрекинг и каталитический крекинг. Из деструктивных процессов переработки нефти наиболее широко используется каталитический крекинг. Большинство процессов каталитического крекинга в мире проводится в "кипящем слое" катализатора.
1.2.1. Каталитический крекинг
Из каталитических процессов наиболее важным является процесс каталитического крекинга нефтяного сырья с получением продуктов меньшей молекулярной массы: компонентов высокооктановых бензинов, легкого газойля, углеводородных газов С3-С4 и др. Приблизительно около одной трети всей добываемой нефти перерабатывается на установках каталитического крекинга.
Процесс каталитического крекинга впервые был осуществлен в промышленном масштабе в 1936 г. для переработки керосино-газойлевых фракций с целью получения бензина. Процесс проводили в стационарном слое катализатора, в качестве которого использовали кислые природные глины - монтмориллониты. В 1940 г. использование в качестве катализатора алюмо-
36
силиката позволило повысить выход бензиновых фракций. В 1941 г. процесс крекинга стали проводить в "кипящем слое" на микросферическом катализаторе, а в 1942 г. - в движущемся слое гранулированного шарикового катализатора.
Значительное улучшение процесса каталитического крекинга связано с открытием каталитической активности цеолитов и цеолитсодержащих катализаторов на их основе. Впервые цеолиты применили в промышленном масштабе в 1962 г. Их использование позволило осуществить крекинг в лифтреакторе.
При проведении каталитического крекинга протекает ряд последова- тельно-параллельных реакций, в которых участвуют как молекулы исходных веществ, так и вещества, образующиеся в ходе реакции:
-крекинг насыщенных углеводородов с образованием алифатических углеводородов меньшей молекулярной массы;
-крекинг циклических углеводородов с образованием алкенов ( олефинов);
-деалкилирование алкилароматических углеводородов;
-расщепление боковых цепей алкилароматических углеводородов;
-крекинг олефинов с образованием олефинов меньшей молекулярной массы;
-изомеризация;
-диспропорционирование олефинов с низкой молекулярной массой;
-перераспределение водорода;
-полимеризация, конденсация и коксообразование.
При каталитическом крекинге насыщенных углеводородов образуются менее высокомолекулярные алканы и олефины, так как чем больше молекулярная масса углеводорода, тем легче идет расщепление. Крекинг алканов нормального строения сопровождается вторичными реакциями, приводящими к образованию ароматических углеводородов и кокса. Нафтеновые углеводороды с длинными алкильными цепями превращаются в алкилнафтеновые или алкилароматические углеводороды со сравнительно короткими боковыми цепями. Крекинг ароматических углеводородов, преимущественно алкилароматических, сопровождается их деалкилированием и переалкилированием, а также конденсацией. При деалкилировании образуются алканы, олефины и алкилароматические соединения меньшей молекулярной массы. Конденсация ароматических углеводородов друг с другом или с непредельными соединениями приводит к образованию полициклических углеводородов, что способствует отложению кокса на поверхности катализатора. Этот кокс состоит из агломератов с псевдографитовой структурой и многоядерных ароматических углеводородов. С повышением глубины конверсии сырья, температуры реакции и длительности контакта катализатора с сырьем увеличивается соотношение углерод:водород в коксе.

37
Расщепление связей С—С протекает по ионному (гетеролитическому) механизму. Катализаторами гетеролитического расщепления связей С—С являются алюмосиликаты. Катализатор в этом процессе - донор протонов Н+, который можно условно обозначить Н+А-. Ион водорода при присоединении к олефину образует ион СН+, что является начальной стадией ионной цепной реакции распада. Обычно разрыв связи С—С происходит по - правилу: разрывается связь, находящаяся в -положении по отношению к положительно заряженному атому углерода. Такое расщепление приводит к образованию третичного карбкатиона. Например, диизобутилен подвергается следующим превращениям: происходят следующие превращения:
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
CH3К |
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
CH3 C CH2 |
|
C=CH2 + H+A- |
|
CH3 C CH2 |
|
C CH3 + A- ; |
||||||||||||||||||||
|
|
|
|
|
||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||
|
|
|
CH3 CH3 |
|
|
CH3 |
CH3 |
|
||||||||||||||||||||
|
|
|
|
|
|
|
CH3 |
+ |
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
CH3 C CH2 C CH3 |
CH3 C CH3 + CH2=C CH3 ; |
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
CH3 |
|
|
CH3 |
|
CH3 |
|
|
CH3 |
|
||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||||||||||||
+ |
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
CH3 |
+ |
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
СH3 C CH3 |
+ CH3 C CH2 C=CH2 |
CH3 C CH2 C CH3 + СН2=C CH3 |
||||||||||||||||||||||||||
C |
|
CH3 |
|
|
|
|
CH3 |
|
|
С |
|
H3 |
|
|
|
|
CH3 |
|
|
CH3 |
CH3 ; |
|||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
CH3 |
+ |
|
|
|
+ |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
CH3 C CH2 C CH3 |
|
CH3 |
C CH3 |
+ CH2=C CH3 и т.д. |
|||||||||||||||||||||
|
|
|
|
|
|
|
CH3 |
|
|
CH3 |
|
|
CH3 |
|
|
|
CH3 |
|
||||||||||
Обрыв цепи происходит в результате соединения иона карбкатиона с |
||||||||||||||||||||||||||||
анионом катализатора: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
C(CH3)3 + A- |
CH2=C |
CH3 + H+A- |
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
||||
От образующегося третичного карбкатиона отщепляется в данном слу- |
||||||||||||||||||||||||||||
чае молекула олефина с четырьмя атомами углерода – изобутилен: |
|
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
CH3 |
+ |
|
|
|
|
+ |
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
CH3 C CH2 C CH3 |
|
CH3 C CH3 |
+ CH2=C CH3 |
||||||||||||||||||||||
|
|
|
|
|
|
|
CH3 |
|
|
CH3 |
|
|
CH3 |
|
|
|
CH3 |
|
||||||||||
От вторичного карбкатиона отщепляется молекула олефина с тремя уг- |
||||||||||||||||||||||||||||
леродными атомами, например пропилен: |
+ |
|
|
|
|
|
||||||||||||||||||||||
СH2=CHCH2CH2CH2CH2СH3 + H+A- |
|
|
|
|
|
|
||||||||||||||||||||||
|
CH3CHCH2CH2CH2CH2СH3 + A- |
|||||||||||||||||||||||||||
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
||||
CH3CHCH2CH2CH2CH2СH3 |
CH2=CH CH3 + CH2 CH2 CH2 СH3 |
|||||||||||||||||||||||||||
38
Насыщенные углеводороды непосредственно не образуют ионов карбкатионов. По-видимому, сначала они претерпевают термическое превращение в алкан и олефин, например:
CH3 CH2 CH2 CH3  CH3 CH3 + CH2=CH2
CH3 CH3 + CH2=CH2
Образующийся олефин с большой скоростью превращается в ион карбкатиона, который начинает реакционную цепь.
Деструкция циклоалканов протекает аналогично деструкции насыщенных ациклических углеводородов, при этом расщеплению подвергается как цикл, так и боковые цепи. Циклоалканы могут расщепляться по нескольким схемам:
|
Алкилциклоалкан |
||
|
↓ |
Алкан+циклоолефин |
|
Олефин +олефин |
↓ |
||
|
|||
↓
Олефин+циклоалкан
Бензол и замещенные бензолы при каталитическом процессе не расщепляются. В замещенных бензолах первичное расщепление происходит по месту присоединения заместителя к кольцу:

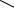
 CH2-CH2-CH3
CH2-CH2-CH3
+ CH3 CH=CH2
Тепловые эффекты реакций каталитического крекинга различаются по величине и знаку. Реакции разрыва С—С-связей протекают с поглощением тепла, а реакции изомеризации, циклизации, полимеризации олефинов, алкилирования, гидрирования и некоторые другие - с выделением тепла. Преобладающими являются эндотермические реакции.
Количество продуктов крекинга и их состав зависят от характера сырья, природы катализатора и технологических параметров процесса. Температура процесса определяется характеристиками катализатора и сырья, временем их контакта, назначением процесса, конструкцией реакторного блока. Реальный температурный интервал процесса каталитического крекинга 723803 К. С повышением температуры возрастает степень конверсии, увеличивается выход сухого газа и бутан-бутиленовой фракции, уменьшается выход бензина, усиливается коксообразование, увеличивается степень ароматизации продуктов крекинга, что приводит к повышению октанового числа бензина крекинга и снижению цетанового числа компонентов дизельной фракции. В бензиновых фракциях с повышением температуры снижается содержание насыщенных углеводородов и увеличивается содержание олефинов.
Первоначально каталитический крекинг проводили при давлении в реакторе и регенераторе не более 0,07 МПа. В дальнейшем для интенсификации выжига кокса с поверхности катализатора давление увеличили до 0,4 МПа. Поскольку с повышением давления интенсивность коксообразования
39
значительно возрастает, то для уменьшения выхода кокса сырье разбавляют водяным паром. Это особенно важно при переработке тяжелого сырья.
Степень превращения сырья и выход продуктов в значительной степени определяются временем контакта сырья с катализатором. Чем меньше продолжительность пребывания сырья в реакционной зоне, тем ниже степень его конверсии. Уменьшение времени контакта может быть скомпенсировано более высокой активностью катализатора и повышением температуры.
Сырье и целевые продукты
В качестве сырья в процессах каталитического крекинга используют керосино-газойлевые фракции (473-623 К) и вакуумные газойли (623-773 К).
Присутствие в сырье металлов, коксообразующих веществ, азотистых и сернистых соединений отрицательно влияет на эффективность каталитического крекинга.
Металлы (никель, ванадий, медь, железо и другие содержатся в высококипящих фракциях в основном в виде порфириновых комплексов. Для устранения их дезактивирующего действия на катализатор крекинга обычно используют два приема:
-выделение металлов из сырья каталитического крекинга - деметаллиза-
ция;
-пассивация металлов, отложившихся на катализаторе.
Суммарное содержание никеля, ванадия и натрия в сырье не должно превышать 1 мас. ч. на 1млн.
Один из самых рациональных способов подготовки нефтяных фракций для каталитического крекинга - гидроочистка, позволяющая значительно повысить степень превращения сырья и выход бензина, а также понизить коксообразование и содержание серы в продуктах крекинга.
Катализаторы процесса крекинга
Первые установки каталитического крекинга с неподвижным слоем катализатора, в качестве которого использовали кислые природные глины, появились в 1940-х годах. В 1950-х годах вместо катализаторов на основе природных глин стали применять аморфные синтетические алюмосиликаты. Эти катализаторы значительно активнее, чем катализаторы на основе глин; обладают большей стабильностью и механической прочностью. В середине 1960- х годов появились аморфные алюмосиликатные катализаторы с высоким со-
держанием Аl2О3.
Одновременно были разработаны цеолитные катализаторы крекинга, которые оказались на несколько порядков активнее аморфных алюмосиликатных катализаторов и позволяли значительно увеличить выход бензина.
40
Создание цеолитных катализаторов крекинга поистине совершило переворот в нефтепереработке, прежде всего в процессах каталитического крекинга.
Цеолиты представляют собой алюмосиликаты каркасной структуры с полостями, занятыми большими ионами и молекулами воды, которые характеризуются значительной подвижностью. Это обеспечивает возможность ионного обмена и обратимой дегидратации.
Совершенствование цеолитных катализаторов привело к созданию новой технологии каталитического крекинга – к процессу крекинга с подвижным потоком пылевидного катализатора, это так называемый крекинг в лифтреакторе. Технология была реализована в начале 1980-х годов, и в настоящее время крекинг в лифт-реакторе является главным в нефтепереработке.
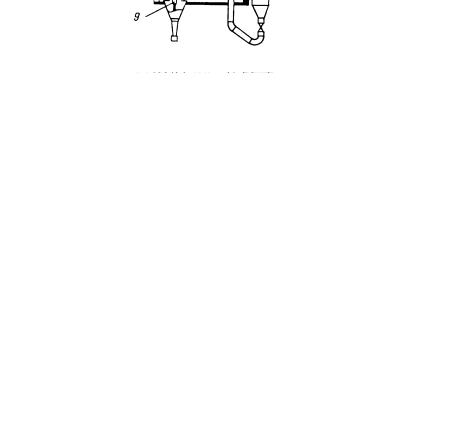
Одними из наиболее распространенных установок каталитического крекинга являются установки фирмы "Келлог". На рис. 1.3 представлен реакторнорегенераторный блок процесса крекинга фирмы "Келлог". Установка позволяет перерабатывать вакуумные газойли и мазуты в высококачественные продукты.
Рис. 1.3. Реакторно-регенеративный блок установки каталитического крекинга по лицензии фирмы "Келлог"
1 - боковой отвод; 2 - расширительная муфта; 3 - вертикальный лифтреактор; 4 - отвод с правым поворотом; 5 - система закрытых циклонов; 6 - двухступенчатый десорбер; 7 - регенератор; 8 - внешний коллектор