

Molecular Fluorescence
.pdf4.5 Photoinduced proton transfer 105
The determination of the energies of the 0–0 transitions is the major cause of inaccuracy in the estimation of pK . In fact, a deviation of 4 nm, e.g. around 400 nm, corresponds to a deviation of one unit on the value of pK .
The Fo¨rster cycle method is quite simple, which explains why it has been extensively used. One of the important features of this cycle is that it can be used even in cases where the equilibrium is not established within the excited-state lifetime. However, use of the Fo¨rster cycle is di cult or questionable when (i) two absorption bands overlap; (ii) the electronic levels invert during the excited-state lifetime (usually in a solvent-assisted relaxation process); (iii) the excited acidic and basic forms are of di erent orbital origins (electronic configuration or state symmetry); and (iv) the changes in dipole moment upon excitation are di erent for the acidic and basic forms.
4.5.2.2 Steady-state measurements
The value of the excited-state pK can be determined by fluorometric titration, but only when the equilibrium is established in the excited state.
In the presence of photoinduced proton transfer, the steady-state fluorescence intensities are given by Eqs (4.55) and (4.56). In the absence of deprotoration (i.e. in a very acidic solution such that k 1½H3Oþ& g1=t00 ), when the experimental conditions (concentrations, excitation and observation wavelengths, sensitivity of the instrument) are kept strictly identical, the fluorescence intensities is ðIAH Þ0 ¼ CF0. Rewriting Eqs (4.55) as IAH ¼ CF, the following ratio is obtained
IAH |
|
F |
|
1 þ k 1t00 ½H3Oþ& |
4:67 |
ðIAH Þ0 ¼ |
|
¼ |
|||
F0 |
1 þ k1t0 þ k 1t00 ½H3Oþ& |
ð Þ |
|||
In the same way, in a su ciently basic medium, ðIA Þ0 ¼ C0F00 , and rewriting eq. 4.56 as IA ¼ C0F00 , we obtain
|
|
IA |
¼ |
|
F0 |
¼ |
|
|
|
|
k1t0 |
|
|
|
|
ð4:68Þ |
|||||
ð |
IA |
Þ0 |
|
F0 |
1 |
þ |
k1t0 |
k |
1t0 |
|
H3Oþ |
& |
|
||||||||
|
|
0 |
|
|
|
|
|
þ |
0½ |
|
|
|
|
||||||||
The ratio of these two equations yields |
|
|
|
|
|||||||||||||||||
|
F=F |
1 |
|
|
k |
t0 |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
0 |
|
|
|
|
|
|
1 |
0 |
H Oþ |
|
|
|
|
|
4:69 |
||
F0 |
=F0 |
¼ k1t0 þ |
& |
|
|
|
|
||||||||||||||
k1t0 ½ |
|
3 |
|
|
|
|
ð |
Þ |
|||||||||||||
|
|
0 |
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Therefore, if |
the |
excited-state |
lifetimes t0 and t00 |
are known, the plot |
of |
||||||||||||||||
ðF=F0Þ=ðF0=F00 Þ versus [H3Oþ] yields the rate constants k1 and k 1. However, it should be emphasized that corrections have to be made: (i) the proton concentration must be replaced by the proton activity; (ii) the rate constant k 1 must be multiplied by a correction factor involving the ionic strength (if the reaction takes place between charged particles), because of the screening e ect of the ionic atmosphere on the charged reactive species.

106 4 Effects of intermolecular photophysical processes on fluorescence emission
4.5.2.3 Time-resolved experiments
The most reliable method for the determination of k1 and k 1 is based on timeresolved experiments. Either pulse fluorometry or phase fluorometry can be used (see Chapter 6). They provide the values of the decay times from which the rate constants k1 and k 1 are determined from Eqs (4.52) to (4.53) and the ratio k1=k 1 yields K .
In some cases, a long tail can be detected in the decay. It has been assigned to geminate recombination according to the following kinetic scheme:
AH þ H2O 8 A . . . H3Oþ ! A þ H3Oþ
k 1ðtÞ
The ions recombine before their mutual distance is greater than the radius of the Coulomb cage. This is in particular the case of pyranine (see formula in Table 4.4) whose acidic form bears 3 negative charges and the basic form 4 negative charges, which results in a large Coulomb cage.
The rate constant for recombination k 1 is time-dependent and can be approximated to t 3=2 at long times. The geminate recombination explains the residual fluorescence intensity of AH despite the fact that pH gpK (see Section 4.5.3). In restricted media, the tail is even longer because of the higher probability of recapturing a photoejected proton by geminate recombination (see Box 4.3).
4.5.3
pH dependence of absorption and emission spectra
As a result of the acido–basic properties in the ground and excited states, absorption and fluorescence spectra are pH-dependent. Let us recall that after proton ejection in the excited state, proton back-recombination can occur or not, depending of the pH (see section 4.5.1). For pH values greater than @2, this back reaction does take place. No equilibrium is reached in the excited state. Furthermore, distinciton should be made according to the value of pK .
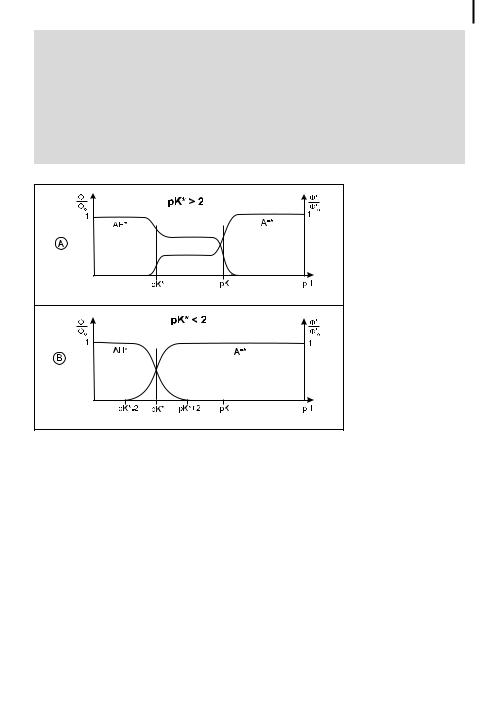
.If pK is greater than @2, a plateau is observed for the relative fluorescence quantum yield of the acidic form and the basic form for pH ranging from pK to pK (Figure 4.10A) because of the absence of di usional recombination. In fact, Eqs (4.59) and (4.60) which are relevant to this case show that IHA and IA are
constants. A typical example is 2-naphthol (pK ¼ 9.3, pK ¼ 2.8).
.If pK is less than @2, the acid is very strong in the excited state and, in general,
k1 is much larger than the reciprocal of the excited-state lifetime, so that the fluorescence of the acidic form is not observed at pH > pK þ 2 but only for lower
pH values (Figure 4.10B). Eqs (4.55) and (4.56) expressing the pH dependence of IHA and IA account for the shape of the curves. However, this is not strictly valid when geminate recombination occurs. A weak emission of the acidic form can then be observed. This is the case for pyranine (pK ¼ 7.7, pK ¼ 1.3) (Figure 4.11).

4.5 Photoinduced proton transfer 107
Box 4.3 Probing the acido–basic properties of water in restricted media or in the vicinity of an interface
The acido–basic properties of water molecules are greatly a ected in restricted media such as the active sites of enzymes, reverse micelles, etc. The ability of water to accept or yield a proton is indeed related to its H-bonded structure which is, in a confined environment, di erent from that of bulk water. Water acidity is then best described by the concept of proton-transfer e ciency – characterized by the rate constants of deprotonation and reprotonation of solutes – instead of the classical concept of pH. Such rate constants can be determined by means of fluorescent acidic or basic probes.
The aqueous cores of reverse micelles are of particular interest because of their analogy with the water pockets in bioaggregates and the active sites of enzymes. Moreover, enzymes solubilized in reverse micelles can exhibit an enhanced catalytic e ciency. Figure B4.3.1 shows a reverse micelle of bis(2- ethylhexyl)sulfosuccinate (AOT) in heptane with three naphthalenic fluorescent probes whose excited-state pK values are much lower than the ground-state pK (see Table 4.4): 2-naphthol (NOH), sodium 2-naphthol sulfonate (NSOH), potassium 2-naphthol-6,8-disulfonate (NSOH). The spectra and the rate constants for deprotonation and back-recombination (determined by time-resolved experiments) provide information on the location of the probes and the corresponding ability of their microenvironment to accept a protona). (i) NDSOH is located around the center of the water pool, and at water contents w ¼ ½H2O&=½AOT& >
Fig. B4.3.1. Schematic illustration of the average residence sites of the probes NOH (1), NSOH (2), NDSOH (3) in AOT reverse micelles. Length of the surfactant: 11 A˚ . Diameter of the water pool: 18 A˚ at w ¼ 3, 36 A˚ at w ¼ 9. Largest dimension of the naphthol derivatives A9 A˚ (adapted from Bardez et al.a)).
108 4 Effects of intermolecular photophysical processes on fluorescence emission
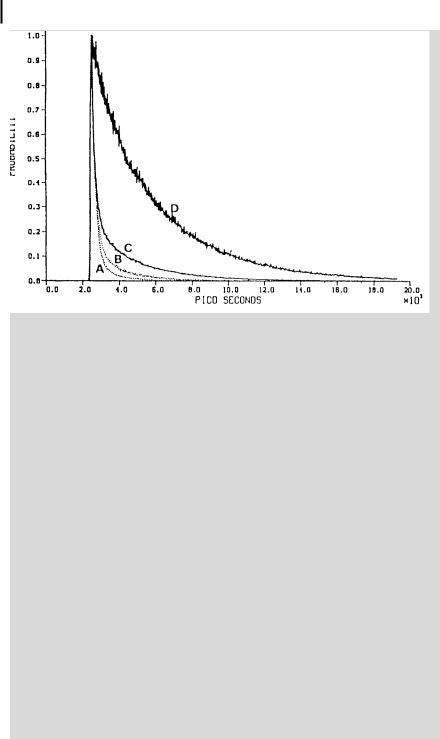
Fig. B4.3.2. Fluorescence decay curves for pyranine in various aqueous environments. See text for the meaning of A, B, C and D (reproduced with permission from Gutman et al.b)).
10, its protolysis behavior is identical to that of bulk water. (ii) NSOH resides in the vicinity of the interface and an amount of water of w A40 is required to observe the same deprotonation rate as in bulk water, whereas the rate of backrecombination is still much faster. (iii) NOH is located at the interface and does not undergo deprotonation in the excited state whatever the water content. E ciency and kinetics of proton transfer are thus strongly dependent on location. The protolytic reactivity of water is related to its H-bonded structure, which changes as a function of the distance with respect to the interface; in the vicinity of the interface, water molecules are in fact involved in the hydration of the polar heads and sodium ions.
Other restricted media have been probed by measuring the fluorescence decay of pyranineb), as shown in Figure B4.3.2:
. Curve A: pyranine in water at pH 5.5;
. Curve B: pyranine trapped in the thin water layer (30 A˚ thickness) of multilamellar vesicles made of dipalmitoylphosphatidyl choline (pH 5.5);
. Curve C: pyranine enclosed in the anion specific pore (18 27 A˚ ) of the PhoE protein;
.Curve D: pyranine in the heme binding site of apomyoglobin, a site containing 30 water molecules or less.
4.6 Excitation energy transfer 109
It can be seen that the smaller the size of the restricted medium, the longer the tail of the fluorescence decay because of the higher probability of recapturing a photoejected proton by geminate recombination. Thus, the fluorescence decay reports the fate of a proton whose life depends on its microenvironment.
a)Bardez E., Monnier E. and Valeur B. (1985)
J. Phys. Chem. 89, 5031–6.
b)Gutman M., Shimoni E. and Tsfadia Y. (1992) Electron and Proton Transfer in
Chemistry and Biology, in: A. Mu¨ller et al. (Eds), Studies in Physical and Theoretical Chemistry, Vol. 78, Elsevier, Amsterdam, pp. 273–85.
Fig. 4.10. Variations in relative fluorescence quantum yields of acidic and basic forms versus pH for various cases.
Fig. 4.11. pH dependence of emission spectra of pyranine in very acidic media. The residual fluorescence of the acidic form at pH > 3 is due to geminate recombination.

1104 Effects of intermolecular photophysical processes on fluorescence emission
4.6
Excitation energy transfer
Energy transfer from an excited molecule (donor) to another that is chemically di erent (acceptor) according to
D þ A ! D þ A
is called heterotransfer. This process is possible provided that the emission spectrum of the donor partially overlaps the absorption spectrum of the acceptor.
If the donor and acceptor are identical, we have homotransfer:
D þ D ! D þ D
When the process can repeat itself so that the excitation migrates over several molecules, it is called excitation transport or energy migration.
4.6.1
Distinction between radiative and non-radiative transfer
It is important to distinguish between radiative and non-radiative transfer. Radiative transfer corresponds to the absorption by a molecule A (or D) of a photon emitted by a molecule D and is observed when the average distance between D and A (or D) is larger than the wavelength. Such a transfer does not require any interaction between the partners, but it depends on the spectral overlap and on the concentration. In contrast, non-radiative transfer occurs without emission of photons at distances less than the wavelength and results from shortor long-range interactions between molecules. For instance, non-radiative transfer by dipole– dipole interaction is possible at distances up to 80–100 A˚ . Consequently, such a transfer provides a tool for determining distances of a few tens of Angstro¨ms between chromophores (see Chapter 9). It is worth noting that the transfer of excitation energy plays a major role in photosynthetic systems.
Radiative and non-radiative transfers have di erent e ects on the characteristics of fluorescence emission from the donor, which allows us to make a distinction between these two types of transfer. Tables 4.5 and 4.6 summarize the e ects of heterotransfer and homotransfer, respectively.
The factors governing the e ciency of radiative and non-radiative transfers are not the same (apart from the spectral overlap, of course, which is required for both processes) (see Table 4.7).
4.6.2
Radiative energy transfer
Radiative transfer is a two-step process: a photon emitted by a donor D is absorbed by an acceptor that is chemically di erent (A) or identical (D)
|
|
|
|
|
4.6 |
Excitation energy transfer |
111 |
||
|
|
||||||||
Tab. 4.5. E ect of energy transfer on the fluorescence characteristics of the donor in the case of |
|||||||||
a heterotransfer (D þ A ! D þ A ) |
|
|
|
|
|
|
|||
Characteristics of the donor |
Radiative transfer |
|
Non-radiative transfer |
||||||
emission |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Fluorescence spectrum |
|
Modified in the region of |
Unchanged |
|
|
|
|||
|
|
|
spectral overlap |
|
|
|
|
|
|
Steady-state fluorescence |
|
Decreased in the region of |
Decreased by the same factor |
||||||
intensity |
|
|
spectral overlap |
|
whatever lem |
|
|
|
|
Fluorescence decay |
|
Unchanged |
|
Shortened |
|
|
|
||
|
|
|
|||||||
Tab. 4.6. E ect of energy transfer on the fluorescence characteristics of the donor in the case of |
|||||||||
a homotransfer (D þ D ! D þ D ) |
|
|
|
|
|
|
|||
Characteristics of the |
|
Radiative transfer |
|
Non-radiative transfer |
|||||
emission |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Fluorescence spectrum |
|
Modified in the region of |
Unchanged |
|
|
|
|||
|
|
|
spectral overlap |
|
|
|
|
|
|
Steady-state fluorescence |
|
Decreased in the region of |
Decreased by the same factor |
||||||
intensity |
|
|
spectral overlap |
|
whatever lem |
|
|
|
|
Fluorescence decay |
|
Slower |
|
Unchanged |
|
|
|
||
Steady-state emission |
|
Decreased |
|
Strongly decreased |
|
|
|
||
anisotropy |
|
|
|
|
|
|
|
|
|
Decay of emission |
|
Faster |
|
Much faster |
|
|
|
||
anisotropy |
|
|
|
|
|
|
|
|
|
see Chapter 5. |
|
|
|
|
|
|
|
|
|
Tab. 4.7. Factors governing the e ciency of radiative and non-radiative transfers |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
Spectral |
lexc and lem |
FD |
rDA or rDD |
Viscosity |
Geometry |
|||
|
overlap |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Radiative transfer |
yes |
|
yes |
no |
no |
no |
yes |
||
Non-radiative transfer |
yes |
|
no |
yes |
yes |
yes |
no |
||
|
|
|
|
|
|
|
|
|
|
FD: fluorescence quantum yield of the donor; rDA; rDD: intermolecular distances.
D ! D þ hn
hn þ A ! A or hn þ D ! D
This process is often called trivial transfer because of the simplicity of the phenomenon, but in reality the quantitative description is quite complicated because it depends on the size of the sample and its configuration with respect to excitation and observation.

112 4 Effects of intermolecular photophysical processes on fluorescence emission
Radiative transfer results in a decrease of the donor fluorescence intensity in the region of spectral overlap. Such a distortion of the fluorescence spectrum is called the inner filter e ect (see Chapter 6).
The fraction a of photons emitted by D and absorbed by A is given by
|
1 |
y |
|
|
a ¼ |
|
ð0 |
IDðlÞ½1 10 eAðlÞCAl& dl |
ð4:71Þ |
FD0 |
where CA is the molar concentration of acceptor, FD0 is the fluorescence quantum yield in the absence of acceptor, l is the thickness of the sample, IDðlÞ and eAðlÞ are
the donor fluorescence intensity and molar absorption coe cient of the acceptor,
Ðy
respectively, with the normalization condition 0 IDðlÞ dl ¼ 1. If the optical density is not too large, a can be approximated by
|
2:3 |
y |
|
|
|
a ¼ |
CAl ð0 |
IDðlÞeAðlÞ dl |
ð4:72Þ |
||
FD0 |
where the integral represents the overlap between the donor fluorescence spectrum and the acceptor absorption spectrum.
In the case of radiative transfer between identical molecules, the fluorescence decays more slowly as a result of successive re-absorptions and re-emissions. A simple kinetic model has been proposed by Birks (1970). It is based on the assumption of a unique value for the average probability a that an emitted photon is absorbed, i.e. without distinction between the generations of photons (a photon of generation n is emitted after n successive re-absorptions). This model leads to the following expressions for the e ective lifetime and the macroscopic fluorescence quantum yield:
t ¼ |
|
t0 |
ð4:73Þ |
|||||||||
1 |
|
F0 |
|
|||||||||
a |
|
|||||||||||
|
|
|
F0 |
|
|
F0 |
|
|
||||
F |
|
|
a |
|
4:74 |
|||||||
|
¼ |
1 |
a |
F0 |
ð |
Þ |
||||||
where t0 and F0 are the lifetime and fluorescence quantum yield, respectively, in the absence of transfer (i.e. as measured at high dilution). This model provides a simple and fast way of estimating the magnitude of radiative transfer under normal conditions of observation of fluorescence. However, according to this model, the fluorescence decay is expected to be a single exponential and does not depend on wavelength, which is not true in practice. It cannot be assumed that photons of di erent generations and di erent wavelengths have the same probability of being absorbed.
Stochastic approaches and Monte Carlo simulations o er a better description of radiative transfer, as shown by Berberan-Santos and coworkers (1999).
4.6 Excitation energy transfer 113
4.6.3
Non-radiative energy transfer
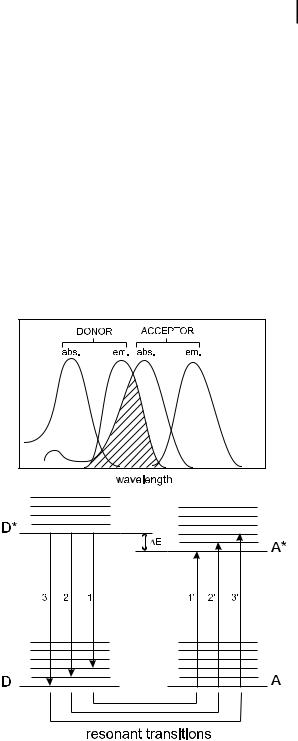
Non-radiative transfer of excitation energy requires some interaction between a donor molecule and an acceptor molecule, and it can occur if the emission spectrum of the donor overlaps the absorption spectrum of the acceptor, so that several vibronic transitions in the donor have practically the same energy as the corresponding transitions in the acceptor. Such transitions are coupled (see Figure 4.12), i.e. are in resonance. The term resonance energy transfer (RET) is often used. In some papers, the acronym FRET is used, denoting fluorescence resonance energy transfer, but this expression is incorrect because it is not the fluorescence that is transferred but the electronic energy of the donor. Therefore, it is recommended that either EET (excitation energy transfer or electronic energy transfer) or RET (resonance energy transfer) are used.
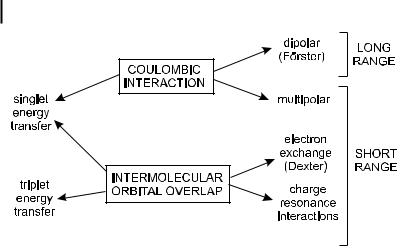
Energy transfer can result from di erent interaction mechanisms. The interactions may be Coulombic and/or due to intermolecular orbital overlap. The Coulombic interactions consist of long-range dipole–dipole interactions (Fo¨rster’s
Fig. 4.12. Energy level scheme of donor and acceptor molecules showing the coupled transitions in the case where vibrational relaxation is faster than energy transfer (very weak coupling) and illustration of the integral overlap between the emission spectrum of the donor and the absorption of the acceptor.
114 4 Effects of intermolecular photophysical processes on fluorescence emission
Fig. 4.13. Types of interactions involved in non-radiative transfer mechanisms.
mechanism) and short-range multi-polar interactions. The interactions due to intermolecular orbital overlap, which include electron exchange (Dexter’s mechanism) and charge resonance interactions, are of course only short range (Figure 4.13). It should be noted that for singlet–singlet energy transfer ð1D þ 1A ! 1D þ 1A Þ, all types of interactions are involved, whereas triplet–triplet energy transfer ð3D þ 1A ! 1D þ 3A Þ is due only to orbital overlap. The reason will be given later.
Box 4.4 shows how the total interaction energy can be expressed as a sum of two terms: a Coulombic term Uc and an exchange term Uex. The Coulombic term corresponds to the energy transfer process in which the initially excited electron on the donor D returns to the ground state orbital on D, while simultaneously an electron on the acceptor A is promoted to the excited state (Figure 4.14A). The exchange term (which has a quantum-mechanical origin) corresponds to an energy transfer process associated with an exchange of two electrons between D and A (Figure 4.14B).
For allowed transitions on D and A the Coulombic interaction is predominant, even at short distances. For forbidden transitions on D and A (e.g. in the case of transfer between triplet states ð3D þ 1A ! 1D þ 3A Þ, in which the transitions T1 ! S0 in D and S0 ! T1 in A are forbidden), the Coulombic interaction is negligible and the exchange mechanism is found, but is operative only at short distances (< 10 A˚ ) because it requires overlap of the molecular orbitals. In contrast, the Coulombic mechanism can still be e ective at large distances (up to 80–100 A˚ ).
The magnitude of the interaction is even more important than its nature, and it is thus convenient to make a distinction, as proposed by Fo¨rster, between three main classes of coupling (strong, weak and very weak), depending on the relative values of the interaction energy (U), the electronic energy di erence between D and A ðDEÞ, the absorption bandwidth ðDwÞ and the vibronic bandwidth ðDeÞ (Figure 4.15).