

Molecular Fluorescence
.pdf3.4 Effects of molecular structure on fluorescence 65
separation) are exceptional. The existence of TICT states (fluorescent or not) has often been invoked (sometimes abusively!) to interpret the photophysical properties of fluorophores, especially those possessing an anilino moiety. When there are several possibilities for internal rotation in an excited molecule, interpretation of the photophysical properties becomes di cult. For instance, in the case of DCM (a well-known merocyanine widely used as a laser dye), several rotations about single bonds are possible. Moreover, photoisomerization (twisting about the double bond) can occur; this process is the main non-radiative de-excitation pathway in nonpolar solvents but it is quite ine cient in polar solvents where the ICT state is stabilized. A very e cient ICT can indeed occur from the dimethylamino group to the dicyanomethylene group, as revealed by the much higer dipole moment of DCM in the excited state than in the ground state (the di erence is about 20 D). Donor–acceptor stilbenes such as DCS exhibit a similar behavior.
Intramolecular charge transfer and internal rotation can also occur in nonpolar and highly symmetric molecules. An outstanding example is 9,90-bianthryl. This compound in fact exhibits dual fluorescence and the band located at higher wavelengths exhibits a large red-shift as the solvent polarity increases, which is indicative of a highly dipolar character (TICT state). Thus, the high symmetry of bianthryl is broken when the charge transfer state is formed.
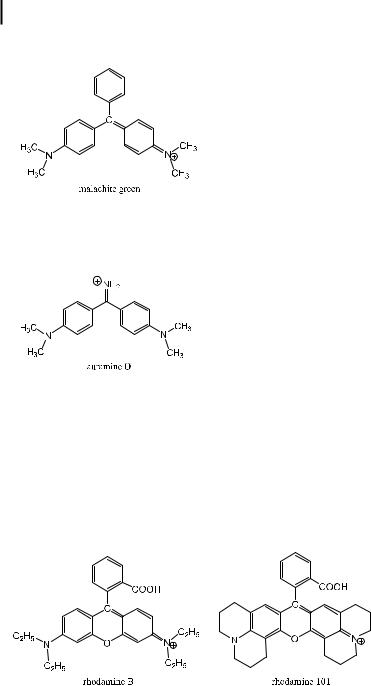
Triphenylmethane dyes such as malachite green are short-chain cyanine dyes possessing two equivalent resonance structures with a charged and an uncharged nitrogen atom. Consequently, they are symmetric with respect to charge distribution. The photophysics of these dyes is quite complex. Their fluorescence quantum yields and decays are very sensitive to the solvent viscosity, which can be explained by the rotational di usion of the phenyl rings occurring along a barrierless potential, with a non-radiative decay rate that depends on the twist angle. The fluorescence quantum yield is thus dependent on solvent viscosity but the internal rota-
66 3 Characteristics of fluorescence emission
tions do not reflect the macroscopic viscosity of the solvent because the free volume e ects are important. This point will be discussed in Chapter 8.
The substituted diphenylmethane dye, auramine O, is weakly fluorescent in fluid solvents but highly fluorescent in viscous or rigid media. It was originally used to probe the viscosity of viscous polymeric samples. Such a strong dependence on solvent viscosity can be explained in the same way as for triphenylmethane dyes.
As a general rule, internal rotations often provide additional channels for nonradiative de-excitation. Bridging the triphenylmethane structure with an oxygen atom reduces the possibilities of internal rotation. The resulting compounds are rhodamines with much higher fluorescence quantum yields (see Section 3.4.3). Further reduction of internal rotation can be achieved via inclusion of the nitrogen atoms in a julolidyl ring, e.g. in rhodamine 101. Consequently, the fluorescence quantum yield of rhodamine 101 is higher (0.92 in ethanol) than that of rhodamine B (0.54 in ethanol)11). Moreover, in contrast to rhodamine B, the fluorescence quantum yield of rhodamine 101 is almost independent of temperature, and so it is preferred as a quantum counter in spectrofluorometers.
11)These compounds are sensitive to pH because of the presence of the carboxylic group. The given fluorescence quantum
yields are those of the acidic form. There is of course no pH sensitivity exhibited by the ester derivatives (e.g. rhodamine 6G).
3.5 Environmental factors affecting fluorescence 67
Rhodamine di ers from malachite green in the same way as fluorescein di ers from phenolphthalein. The latter is known to be non-fluorescent.
The photophysics of fluorophores undergoing photoinduced charge transfer and/or internal rotation(s) is often complex. Time-resolved fluorescence experiments, transient absorption spectroscopy measurements, quantum chemical calculations, and comparison with model molecules are helpful in understanding their complex photophysical behavior.
3.5
Environmental factors a ecting fluorescence
In Section 3.4, structural e ects were often discussed in conjunction with the nature of the solvent. As emphasized in the introduction to this book, the fluorescence emitted by most molecules is indeed extremely sensitive to their microenvironment (see Figure 1.3), which explains the extensive use of fluorescent probes. The e ects of solvent polarity, viscosity and acidity deserves much attention because these e ects are the basis of fluorescence probing of these microenvironmental characteristics and so, later chapters of this book are devoted to these aspects. The e ects of polarity and viscosity on fluorescence characteristics in fluid media and the relevant applications are presented in Chapters 7 and 8, respectively. The e ect of acidity is discussed in Sections 4.5 and 10.2. This section is thus mainly devoted to rigid matrices or very viscous media, and gases.
3.5.1
Homogeneous and inhomogeneous band broadening. Red-edge e ects
The width of a band in the absorption or emission spectrum of a fluorophore located in a particular microenvironment is a result of two e ects: homogeneous and inhomogeneous broadening. Homogeneous broadening is due to the existence of a continuous set of vibrational sublevels in each electronic state. Absorption and emission spectra of moderately large and rigid fluorophores in solution could therefore be almost structureless at room temperature. However, in some cases, many of the vibrational modes are not active, neither in absorption nor in emission, so that a clear vibrational structure is observed (e.g. naphthalene, pyrene).

68 3 Characteristics of fluorescence emission
The second cause of broadening of electronic spectra is the fluctuations in the structure of the solvation shell surrounding the fluorophore. The distribution of solute–solvent configurations and the consequent variation in the local electric field leads to a statistical distribution of the energies of the electronic transitions. This phenomenon is called inhomogeneous broadening (for a review see Nemkovich et al., 1991).
In most cases, the extent of inhomogeneous broadening is much greater than that of homogeneous broadening. When interactions with the surrounding molecules are strong, and many configurations are possible, the spectra may become completely blurred. Such spectra are of limited value in analytical fluorescence spectroscopy, especially when the samples contain several compounds whose fluorescence spectra overlap. However, there are several ways to reduce the e ect of inhomogeneous broadening for analytical purposes (see Section 3.5.2).
In polar rigid media such as frozen solutions or polymer matrices, inhomogeneous broadening is reduced but still exists, and is responsible for red-edge e ects, i.e. specific e ects that are observed when the fluorophores are excited on the rededge of the absorption spectrum. Red-edge excitation selects the ‘hot’ molecules, i.e. those that absorb from vibrational levels above that of the lowest vibrational level of the ground state. The corresponding fluorescence spectrum is red-shifted with respect to the fluorescence spectrum observed upon excitation in the bulk of the absorption spectrum. In liquid solutions, the inhomogeneous broadening becomes dynamic, and the red-shift disappears because a dynamic equilibrium exists among the various solvation sites, but it is still observable if the solvent reorientation relaxation competes with the fluorescence decay.
An excitation-wavelength dependence at the longwave edge of the absorption spectrum has been observed not only for spectral displacement but also for other parameters such as lifetime, quantum yield and apparent rotational rate. Applications to the investigation of polymer rigidity and/or free volume, and to the study of biological systems and excited-state reactions have been developed.
Finally, there is a specific red-edge e ect related to non-radiative energy transfer between a donor fluorophore whose emission spectrum overlaps the absorption spectrum of an acceptor fluorophore: in rigid polar solutions, there is a lack of energy transfer upon excitation at the red-edge. This e ect, called Weber’s e ect, will be described in Section 9.4.3.
3.5.2
Solid matrices at low temperature
For applications to chemical analysis, a high spectral resolution is desirable and can be achieved using solid matrices at low temperature. In fact, under these conditions, the degree of microenvironmental heterogeneity (expressed as the number of solvation sub-classes) is reduced, which results in decreased inhomogeneous broadening. Moreover, it is advantageous to use laser excitation whose linewidth is less than the width of the inhomogeneously broadened absorption band. Selective excitation of individual compounds in complex mixtures is then possible (Wehry and Mamantov, 1981; Hofstraat et al., 1988).
3.5 Environmental factors affecting fluorescence 69
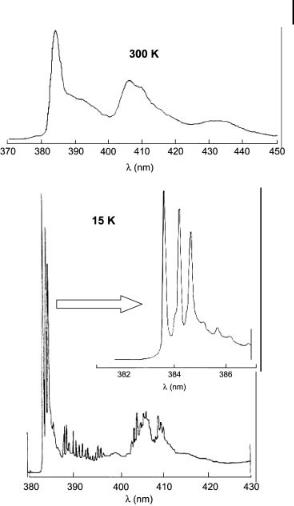
Fig. 3.5. Fluorescence spectrum of benz[a]anthracene in n-heptane liquid solution at 300 K (top) and in n-heptane frozen solution at 15 K (bottom). Concentration:
2 10 5 M. Excitation wavelength: 329.2 nm (adapted from Wehry and Mamantov, 1981).
Shpol’skii spectroscopy Shpol’skii matrices are formed by freezing a solution of fluorophores in n-alkanes (provided that the solubility in these solvents is high enough). Rapid immersion in liquid nitrogen or helium leads to a polycrystalline solid matrix. The solute molecules are in very similar microenvironments and inhomogeneous broadening is therefore considerably reduced (Figure 3.5). The spectral lines are thus very narrow (A10 cm 1 or less) and Shpol’skii spectra can serve as ‘fingerprints’ of complex mixtures. Identification of trace amounts of polynuclear aromatic hydrocarbons (PAHs) in complex samples is a great success of this technique.
Matrix isolation spectroscopy In this technique, the fluorophore in the solid or liquid form is vaporized, and the resulting gaseous sample is mixed with a large excess of an inert gas (rare gases – mainly argon and in some cases xenon or krypton or nitrogen). The mixture is deposited on a solid surface at cryogenic temperatures (15 K or less) obtained with a helium bath cryostat. Aggregation e ects are avoided in such matrices, in contrast to solid samples prepared by freezing of liquid solutions.

70 3 Characteristics of fluorescence emission
In argon or nitrogen matrices, the spectral resolution (bandwidth A100 cm 1) is not as high as in Shpol’skii frozen solutions (bandwidth A10 cm 1 or less). In the case of complex mixtures requiring high resolution, it is thus preferable to use Shpol’skii solvents (n-alkanes and n-perfluoroalkanes).
Site-selection spectroscopy Maximum selectivity in frozen solutions or vapordeposited matrices is achieved by using exciting light whose bandwidth (0.01–0.1 cm 1) is less than that of the inhomogeneously broadened absorption band. Lasers are optimal in this respect. The spectral bandwidths can then be minimized by selective excitation only of those fluorophores that are located in very similar matrix sites. The temperature should be very low (5 K or less). The techniques based on this principle are called in the literature site-selection spectroscopy, fluorescence line narrowing or energy-selection spectroscopy. The solvent (3-methylpentane, ethanol–methanol mixtures, EPA (mixture of ethanol, isopentane and diethyl ether)) should form a clear glass in order to avoid distortion of the spectrum by scatter from cracks.
Various compounds have been analyzed by site-selection spectroscopy. An interesting application is the identification of loci of damage in DNA resulting from reactions with carcinogens such as polycyclic aromatic hydrocarbons.
3.5.3
Fluorescence in supersonic jets
Although this book is devoted to molecular fluorescence in condensed phases, it is worth mentioning the relevance of fluorescence spectroscopy in supersonic jets (Ito et al., 1988). A gas expanded through an orifice from a high-pressure region into a vacuum is cooled by the well-known Joule–Thomson e ect. During expansion, collisions between the gas molecules lead to a dramatic decrease in their translational velocities. Translational temperatures of 1 K or less can be attained in this way. The supersonic jet technique is an alternative low-temperature approach to the solid-phase methods described in Section 3.5.2; all of them have a common aim of improving the spectral resolution.
In a supersonic jet, the fluorescence spectra are virtually free of environmental perturbations (in contrast to condensed-phase samples) and can thus provide information on isolated solute molecules. Moreover, van der Waals complexes with other solute molecules can be studied, which is of great fundamental interest.
3.6
Bibliography
Birks J. B. (1969) Photophysics of Aromatic |
Hofstraat J. W., Gooijer C. and Velthorst |
Molecules, Wiley, London. |
N. H. (1988) Highly resolved molecular |
Birks J. B. (Ed.) (1973) Organic Molecular |
Luminescence Spectroscopy, In: Schulman |
Photophysics, Wiley, London. |
S. G., Ed., Molecular Luminescence |
Guilbault G. G. (Ed.) (1990) Practical |
Spectroscopy, Part 2, John Wiley and Sons, |
Fluorescence, Marcel Dekker, New York. |
New York. pp. 283–400. |
|
3.6 Bibliography |
71 |
Ito M., Ebata T. and Mikami N. (1988) Laser |
|
|
Separation via Twisted Intramolecular |
||
Spectroscopy of Large Polyatomic Molecules |
Charge Transfer States, Topics Curr. Chem. |
|
in Supersonic Jets, Ann. Rev. Phys. Chem. |
169, 253–299. |
|
39, 123–147. |
Turro N. J. (1978) Modern Molecular |
|
Lippert E., Rettig W., Bonacic-Koutecky V., |
Photochemistry, Benjamin/Cummings, |
|
Heisel F. and Miehe´, J. A. (1987) Photo- |
Menlo Park, CA. |
|
physics of Internal Twisting, Adv. Chem. |
Wehry E. L. (1990) E ects of Molecular |
|
Phys. 68, 1–173. |
Structure on Fluorescence and |
|
Nemkovich N. A., Rubinov A. N. and Tomin |
Phosphorescence, in: Guilbault G. G. |
|
I. T. (1991) Inhomogeneous Broadening of |
(Ed.), Practical Fluorescence, Marcel Dekker, |
|
Electronic Spectra of Dye Molecules in |
New York, pp. 75–125. |
|
Solutions, in: Lakowicz J. R. (Ed.), Topics in |
Wehry E. L. and Mamantov G. (1981) Low- |
|
Fluorescence Spectroscopy, Vol. 2, Principles, |
Temperature Fluorometric Techniques and |
|
Plenum Press, New York, pp. 367–428. |
Their Application to Analytical Chemistry, |
|
Parker C. A. (1968) Photoluminescence of |
in: Wehry E. L. (Ed.), Modern Fluorescence |
|
Solutions, Elsevier, Amsterdam. |
Spectroscopy, Vol. 4, Plenum Press, New |
|
Rettig W. (1994) Photoinduced Charge |
York, pp. 193–250. |
|

Molecular Fluorescence: Principles and Applications. Bernard Valeur
> 2001 Wiley-VCH Verlag GmbH
ISBNs: 3-527-29919-X (Hardcover); 3-527-60024-8 (Electronic)
72
4
E ects of intermolecular photophysical processes on fluorescence emission
Luminescence research has a long history, full of splendor and surprise, and a bright future, promising variegated applications to probe molecules and crystals, to visualize atomic phenomena, . . .
H. J. Queisser, 1981
4.1
Introduction
In Chapter 3 we described the intrinsic pathways of de-excitation of a molecule M ; the sum kM of the rate constants for these processes is equal to the reciprocal of the excited-state lifetime t01):
kM ¼ kr þ kic þ kisc ¼ kr þ knr ¼ 1=t0 |
ð4:1Þ |
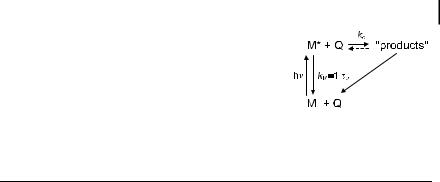
As outlined in Chapter 3, this lifetime is the experimental time window through which processes of similar duration, and competing with the intrinsic de-excitation, can be observed. Most of these processes involve interaction of an excited molecule M with another molecule Q according to Scheme 4.1, where kq represents the observed rate constant for the bimolecular process.
The main intermolecular photophysical processes responsible for de-excitation of molecules are presented in Table 4.1. It is interesting to note that most of them
1)Because this chapter deals only with fluorescence (unless otherwise stated), the subscript F and the superscript S used in Chapter 3 are omitted. The subscript 0 (and
in some cases, the superscript 0) for fluorescence intensity, quantum yield and lifetime will refer to these characteristics in the absence of intermolecular processes.
4.1 Introduction 73
Scheme 4.1
Tab. 4.1. Main photophysical processes responsible for fluorescence quenching
Photophysical process |
M BQ |
! |
products |
|
|
Donor |
Acceptor |
|||||||
|
|
|
||||||||||||
Collision with a heavy atom (e.g., I , Br in |
M |
þ |
Q |
! |
M |
þ |
Q |
þ |
heat |
|
|
|||
CBr4)a) or a paramagnetic species (e.g. |
|
|
|
|
|
|
|
|
||||||
O2, NO)b) |
|
|
|
|
|
|
D.þ |
|
A. |
|
|
|||
Electron transfer |
1D |
þ |
A |
|
|
1D |
A |
|||||||
|
|
|
|
! . |
|
þ . |
þ |
D |
1A |
|||||
|
1A þ D ! A |
þ D |
||||||||||||
Excimer formation |
1M þ 1M ! 1ðMMÞ |
|
|
|||||||||||
Exciplex formation |
1D þ A ! 1ðDAÞ |
|
|
1D |
A |
|||||||||
|
1A þ D ! 1ðDAÞ |
|
|
D |
1A |
|||||||||
Proton transfer |
AH þ B ! A þ BHþ |
AH |
B |
|||||||||||
|
B þ AH ! BHþ þ A |
AH |
B |
|||||||||||
Energy transfer |
1D þ 1A ! 1D þ 1A |
1D |
1A |
|||||||||||
|
3D þ 1A ! 1D þ 3A |
3D |
1A |
|||||||||||
|
3D þ 3A ! 1D þ 3A |
3D |
3A |
|||||||||||
|
1M þ 1M ! 1M þ 1M |
1M |
1M |
|||||||||||
a)As in the case of the intramolecular heavy atom e ect (see Chapter 3), intersystem crossing is favored by collision with heavy atoms.
b)Molecular oxygen is a well-known quencher of fluorescence. It deserves special attention because it is ubiquitous in solutions. Its
ground state is a triplet state (denoted 3O2 or 3S) and it has two low-lying singlet states (1D and 1S). Quenching of singlet or triplet states via energy transfer to produce 1D oxygen is thus possible:
1A þ 3O2ð3SÞ ! 1A þ 1O2ð1DÞ or
3A þ 3O2ð3SÞ ! 3A þ 1O2ð1DÞ
‘Chemical’ quenching by formation of a complex A O2 may also be involved in oxygen quenching.
The contribution of oxygen quenching to the decay of an excited state can be expressed by a quenching term kq½O2& to be added to the rate constants of de-excitation. Under atmospheric pressure, the concentration of oxygen in most solvents is 10 3 – 10 4 mol L 1. Therefore, if kq @1010 L mol 1 s 1, the lifetime of A in the presence of O2 cannot be more than 10 6 –10 7 s in airsaturated solutions. The longer the lifetime in the absence of O2, the stronger the sensitivity to the presence of oxygen, and for some applications, solutions must be degassed by bubbling with nitrogen (or argon), or by the freeze–pump–thaw technique.

74 4 Effects of intermolecular photophysical processes on fluorescence emission
involve a fast transfer process from a donor to an acceptor: electron transfer, proton transfer or energy transfer.
The fluorescence characteristics (decay time and/or fluorescence quantum yield) of M are a ected by the presence of Q as a result of competition between the intrinsic de-excitation and these intermolecular processes:
(i)after excitation by a light pulse, the excited-state M population, and consequently the fluorescence intensity, decreases more rapidly than in the absence of excited-state interaction with Q.
(ii)the fluorescence quantum yield is decreased. The loss of fluorescence intensity is called fluorescence quenching whatever the nature of the competing intermolecular process and even if this process leads to a fluorescent species (the word quenching applies only to the initially excited molecule).
Analysis of the observed phenomena provides much information on the surroundings of a fluorescent molecule, either quantitatively (if a kinetic model of the competitive processes can be developed so that a kinetic analysis is possible), or at least qualitatively. Special attention will be paid in this chapter to the case where fluorescent molecules are used for probing matter or living systems.
This chapter is restricted to intermolecular photophysical processes2). Intramolecular excited-state processes will not be considered here, but it should be noted that they can also a ect the fluorescence characteristics: intramolecular charge transfer, internal rotation (e.g. formation of twisted charge transfer states), intramolecular proton transfer, etc.
Photochemical de-excitation (i.e. de-excitation resulting from organic photochemical reactions implying bond breaking and formation of new bonds so that the ground state of M is not recovered) is beyond the scope of this book.
4.2
Overview of the intermolecular de-excitation processes of excited molecules leading to fluorescence quenching
4.2.1
Phenomenological approach
Several cases can be identified in which an intermolecular process involving M and Q is in competition with intrinsic de-excitation of M .
.Case A: Q is in large excess so that there is a high probability that M and Q, at the time of excitation, are at a distance where the interaction is significant. Then, no mutual approach during the excited-state lifetime is required.
2)The word ‘photophysical’ implies that, after completion of all de-excitation processes, the ground state of M is recovered unaltered.