

- •VOLUME 5
- •CONTRIBUTOR LIST
- •PREFACE
- •LIST OF ARTICLES
- •ABBREVIATIONS AND ACRONYMS
- •CONVERSION FACTORS AND UNIT SYMBOLS
- •NANOPARTICLES
- •NEONATAL MONITORING
- •NERVE CONDUCTION STUDIES.
- •NEUROLOGICAL MONITORS
- •NEUROMUSCULAR STIMULATION.
- •NEUTRON ACTIVATION ANALYSIS
- •NEUTRON BEAM THERAPY
- •NEUROSTIMULATION.
- •NONIONIZING RADIATION, BIOLOGICAL EFFECTS OF
- •NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY
- •NUCLEAR MEDICINE INSTRUMENTATION
- •NUCLEAR MEDICINE, COMPUTERS IN
- •NUTRITION, PARENTERAL
- •NYSTAGMOGRAPHY.
- •OCULAR FUNDUS REFLECTOMETRY
- •OCULAR MOTILITY RECORDING AND NYSTAGMUS
- •OCULOGRAPHY.
- •OFFICE AUTOMATION SYSTEMS
- •OPTICAL FIBERS IN MEDICINE.
- •OPTICAL SENSORS
- •OPTICAL TWEEZERS
- •ORAL CONTRACEPTIVES.
- •ORTHOPEDIC DEVICES MATERIALS AND DESIGN OF
- •ORTHOPEDICS PROSTHESIS FIXATION FOR
- •ORTHOTICS.
- •OSTEOPOROSIS.
- •OVULATION, DETECTION OF.
- •OXYGEN ANALYZERS
- •OXYGEN SENSORS
- •OXYGEN TOXICITY.
- •PACEMAKERS
- •PAIN SYNDROMES.
- •PANCREAS, ARTIFICIAL
- •PARENTERAL NUTRITION.
- •PERINATAL MONITORING.
- •PERIPHERAL VASCULAR NONINVASIVE MEASUREMENTS
- •PET SCAN.
- •PHANTOM MATERIALS IN RADIOLOGY
- •PHARMACOKINETICS AND PHARMACODYNAMICS
- •PHONOCARDIOGRAPHY
- •PHOTOTHERAPY.
- •PHOTOGRAPHY, MEDICAL
- •PHYSIOLOGICAL SYSTEMS MODELING
- •PICTURE ARCHIVING AND COMMUNICATION SYSTEMS
- •PIEZOELECTRIC SENSORS
- •PLETHYSMOGRAPHY.
- •PNEUMATIC ANTISHOCK GARMENT.
- •PNEUMOTACHOMETERS
- •POLYMERASE CHAIN REACTION
- •POLYMERIC MATERIALS
- •POLYMERS.
- •PRODUCT LIABILITY.
- •PROSTHESES, VISUAL.
- •PROSTHESIS FIXATION, ORTHOPEDIC.
- •POROUS MATERIALS FOR BIOLOGICAL APPLICATIONS
- •POSITRON EMISSION TOMOGRAPHY
- •PROSTATE SEED IMPLANTS
- •PTCA.
- •PULMONARY MECHANICS.
- •PULMONARY PHYSIOLOGY
- •PUMPS, INFUSION.
- •QUALITY CONTROL, X-RAY.
- •QUALITY-OF-LIFE MEASURES, CLINICAL SIGNIFICANCE OF
- •RADIATION DETECTORS.
- •RADIATION DOSIMETRY FOR ONCOLOGY
- •RADIATION DOSIMETRY, THREE-DIMENSIONAL
- •RADIATION, EFFECTS OF.
- •RADIATION PROTECTION INSTRUMENTATION
- •RADIATION THERAPY, INTENSITY MODULATED
- •RADIATION THERAPY SIMULATOR
- •RADIATION THERAPY TREATMENT PLANNING, MONTE CARLO CALCULATIONS IN
- •RADIATION THERAPY, QUALITY ASSURANCE IN
- •RADIATION, ULTRAVIOLET.
- •RADIOACTIVE DECAY.
- •RADIOACTIVE SEED IMPLANTATION.
- •RADIOIMMUNODETECTION.
- •RADIOISOTOPE IMAGING EQUIPMENT.
- •RADIOLOGY INFORMATION SYSTEMS
- •RADIOLOGY, PHANTOM MATERIALS.
- •RADIOMETRY.
- •RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY
- •RADIOPHARMACEUTICAL DOSIMETRY
- •RADIOSURGERY, STEREOTACTIC
- •RADIOTHERAPY ACCESSORIES
RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY |
559 |
18.Zick RG, Olsen J. Voice Recognition software Versus a Traditional Transcription Service for Physician Charting in the ED. Am J Emerg Med 2001;19:295–298.
19.Bramson RT, Bramson RA. Overcoming Obstacles to WorkChanging Technology Such As PACS and Voice Recognition. AJR 2005;184:1727–1730.
20.Hussein R, Schroeter A, Meinzer H-P. DICOM Structured Reporting.RadioGraphics 2004;24(3):891–896.
21.Dolin RH, et al. The HL7 Clinical Document Architecture. JAMIA 2001;8:552–579.
22.Flanders AE, Wiggins RH, Gozum ME. Handheld Computers Radiol. RadioGraphics 2003;23:1035–1047.
See also EQUIPMENT ACQUISITION; PICTURE ARCHIVING AND COMMUNICATION SYSTEMS; RADIOTHERAPY TREATMENT PLANNING, OPTIMIZATION OF;
TELERADIOLOGY.
RADIOLOGY, PHANTOM MATERIALS. See
PHANTOM MATERIALS IN RADIOLOGY.
RADIOMETRY. See THERMOGRAPHY.
RADIONUCLIDE PRODUCTION
AND RADIOACTIVE DECAY
SILVIA S. JURISSON
WILLIAM MILLER
J. DAVID ROBERTSON
University of Missouri
Columbia, Missouri
INTRODUCTION
Radiopharmaceuticals, drugs containing radioactive atoms, are used for diagnostic imaging or therapeutic applications in nuclear medicine, depending on their radioactive emissions. Penetrating radiations (gamma rays or annihilation photons from positron emission) are used for diagnostic applications with gamma cameras, single photon emission computed tomography (SPECT), or positron emission tomography (PET) instrumentation. Diagnostic imaging with gamma emitters became a mainstay of nuclear medicine with the advent of the molybdenum-99/technetium-99m (99Mo/99mTc) generator (the Brookhaven generator), which was developed by Richards in 1961 at Brookhaven National Laboratory (1). This generator made the short half-lived 99mTc (6.01 h; 140 keV g-ray) readily available to nuclear medicine departments around the world, and not just at the site of 99mTc production. Today, 99mTc accounts for >80% of the diagnostic scans performed in nuclear medicine departments in the United States, with a variety of U.S. Food and Drug Administration (FDA) approved agents for functional imaging of the heart, liver, gallbladder, kidneys, brain, and so on (2). Particle emitters, such as alpha, beta, and Auger electron emitters, are used for targeted radiotherapy since their decay energy is deposited over a very short range in tissue. Radiotherapeutic applications in humans began in the late 1930s with the beta emitters radioiodine (128I, 131I), radiophosphorus (32P) and
radiostrontium (89Sr) for cancer treatment (3). Radioiodine (131I iodide) was the first, and continues to be the only, true ‘‘magic bullet’’ through its specific and selective uptake in thyroid. It is used to treat hyperthyroidism and thyroid cancer.
Radionuclides for medical applications are produced either at nuclear reactors or accelerators. The ‘‘Availability of Radioactive Isotopes’’ was first announced from the headquarters of the Manhattan Project (Washington, DC) in Science in 1946 (4), and today medical isotope availability remains an important issue for the nuclear medicine community. The selection of radionuclides for use in radiopharmaceuticals is dependent on their decay properties (half-life, emissions, energies of emissions, dose rates) and their availability (production and cost).
Radionuclides suitable for diagnostic imaging are gamma emitters (with no or minimal accompanying particle emissions) such as 99mTc and positron emitters (annihilation photons) such as 18F. The half-lives should be as short as possible and still allow preparation (synthesis and purification) of the radiopharmaceutical, administration of the agent to the patient, and the diagnostic imaging procedure. Typically, half-lives on the order of minutes to a week are used, with hours to a day considered optimal for most applications. Gamma energies in the range of 80– 300 keV are considered good, although higher energy (e.g., 131I at 364.5 keV) are used, and 100–200 keV is considered optimal. The PET instrumentation is designed for the two 511 keV annihilation photons.
The radionuclides used or under investigation for radiotherapeutic applications are particle emitters, with most
of the efforts focusing on the beta emitters (e.g., 153Sm, 90Y, 186Re, 188Re, 177Lu, 149Pm, 166Ho, 105Rh, 199Au) and some on alpha emitters (e.g., 212Bi, 213Bi, 225Ac). In the case of
radiotherapy, the half-life of the radionuclide should match the biological half-life for the radiopharmaceutical delivery to its in vivo target site (i.e., tumor), typically < 1 day – 1 week. The optimum particle and particle energy remains under investigation, and it is not clear that a higher particle energy translates into a more successful treatment (i.e., radiation dose to nontarget organs will limit the dose allowed for administration). Suitable accompanying gamma emissions will allow the in vivo tracking of the radiotherapeutic dose.
Two important factors for the use of radionuclides in nuclear medicine are thus radioactive decay and radionuclide production, both of which are discussed in detail below. Radionuclides used in nuclear medicine applications are used as examples in discussing these topics. Radioactive decay includes the types of decay (e.g., alpha, beta, electron capture, positron, gamma) with focus on those modes with nuclear medicine applications and rates of radioactive decay (e.g., units of radioactivity, exponential decay, activity–mass relationships, parent–daughter equilibria, medical generators).
TYPES OF RADIOACTIVE DECAY
A nuclide is considered to be radioactive if its nuclear configuration (number of neutrons and protons in the
560 RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY
nucleus) is unstable. Radioactive decay is then a means for the unstable nucleus to achieve a more stable nuclear configuration although it may or may not form a stable nuclide following a single decay. There are several modes of decay possible for a radioactive nuclide including alpha emission, beta emission, positron emission, electron capture decay and/or gamma emission. An alpha (a) particle is a helium nucleus (42He2þ) and is generally a mode of decay available in heavy nuclei (i.e., Z 83). Some lanthanides can show a-decay and heavy nuclides can also undergo decay by b , EC, and spontaneous fission. The following equation gives an example of alpha decay:
213Bi |
45:59 min |
209Tl |
4He2þ |
þ |
g |
þ |
Q |
83 |
! |
81 |
þ 2 |
|
|
where Q is the energy released during the decay process and g is the 727 keV gamma photon emitted in 11.8% of the decays. Most of the decay energy in this process is in the form of the kinetic energy of the a-particle, which is rapidly deposited in matter (e.g., tissue) because of its relatively high charge and mass.
Beta (b ) decay occurs when the nucleus has a proton/neutron ratio that is too low relative to the proton/neutron ratio in the stable nuclei of that element, and during this process a neutron is converted into a proton resulting in a nuclide that is one atomic number (Z) higher than its parent. An example of beta decay is shown below,
188 |
16:98 h |
188 |
0 |
75Re ! |
76Os þ 1b þ ~ne þ g þ Q |
||
where Q is the energy released, n~e is an antineutrino, and g is the 155 keV gamma photon emitted in 15% of the decays. These neutron-rich radionuclides are produced in nuclear reactors as will be discussed in the section Radionuclear Production.
When a radioactive nucleus has a proton/neutron ratio that is too high relative to that of the stable nuclei of the same element, two modes of decay are possible (positron emission and/or electron capture decay), with both converting a proton into a neutron resulting in a nuclide that is one atomic number lower than its parent. Electron capture decay arises from the overlap of nucleon orbitals with electron orbitals. It is a result of the ‘‘weak force’’, which is very short ranged. During electron capture decay (e), essentially an inner-shell electron (usually K-shell) is incorporated into the nucleus converting a proton into a neutron. This process creates an orbital vacancy and results in a cascade of outer-shell electrons filling the lower energy inner-shell vacancies, with the excess energy released as X-rays and/or Auger electrons. The following equation shows the electron capture process:
201 0 73:1 h 201
81Tl þ 1e ! 80Hg þ ne þ Q þ Auger e =X rays
where Q is the energy released and ne is a neutrino. The 80 keV X rays emitted are used for myocardial imaging in stress–rest tests performed in nuclear medicine departments. Positron emission is only possible when Q is > 1.022 MeV, the energy equivalent of the mass of two electrons. During positron decay, a proton is converted into
a neutron with simultaneous emission of a positron (bþ), which is a positive electron. Since a neutron has greater mass than a proton, the energy equivalent of two electrons (one to convert a proton to a neutron in the nucleus, and one emitted as a positron) must be available for this decay mode to be possible. Practically, positron emission does not occur unless Q is 2 MeV. An example of the positron decay process is shown below.
18 110 min 18 0 þ
9F ! 8O þ 1b þ ne þ Q
01eþ þ 01e ! 2 511 keV annihilation photons
where Q is the decay energy (including the maximum positron energy plus 1.022 MeV), ne is a neutrino, and the two 0.511 MeV photons result from positron annihilation (often called annihilation photons) and are emitted 1808 opposite each other. They are the basis of PET imaging. The 18F radiolabeled fluorodeoxyglucose (18F-FDG) is used in nuclear medicine departments for imaging glucose metabolism, such as found in growing tumors. In some cases, both electron capture and positron emission can occur. These proton-rich radionuclides are produced by accelerators which is discussed below.
Gamma (g) emission can accompany any other decay process (i.e., alpha, beta, electron capture, positron decay) or it can occur without any particle emission. In the latter case, it is called isomeric transition (IT) and occurs when a metastable radioisotope [higher energy excited state of a nucleus with a measurable lifetime ( ns)] decays to the ground state (lower energy state of the nucleus). Isomeric transition is accompanied by energy release without any other change occurring in the nucleus (i.e., the number of
protons and neutrons in the nucleus does not change during IT). The decay of 99mTc is an example of an isomeric
transition.
99m |
6:01 h |
99 |
43 Tc ! |
43Tc þ g þ Q |
|
where Q is the decay energy and g is the 140 keV photon released from the nucleus. The 99mTc in various chemical formulations is used routinely for diagnostic imaging of a variety of diseases and/or organ functions.
Whenever a gamma photon is released from the nucleus, the emission of conversion electrons is also possible. A conversion electron is the emission of an electron that has the energy of the gamma photon minus the electronic atomic binding energy, and it is emitted in place of the gamma photon. The probability of conversion electron emission rather than gamma emission increases as the energy of the gamma photon decreases and it increases with increasing nuclear charge. For example, the 140 keV gamma photon of 99mTc is emitted with 89% abundance;
conversion electrons, rather than gamma photons, are emitted 11% of the time when 99mTc decays to 99Tc.
There are radionuclides in which more than one type of radioactive decay occurs. An example is the decay of 64Cu, which undergoes beta decay (39% of the time), positron emission decay (19% of the time), and electron capture decay (42% of the time) with a weighted average half-life of 12.7 h (the specific half-lives are shown below). This radionuclide has both diagnostic imaging (bþ emission via the
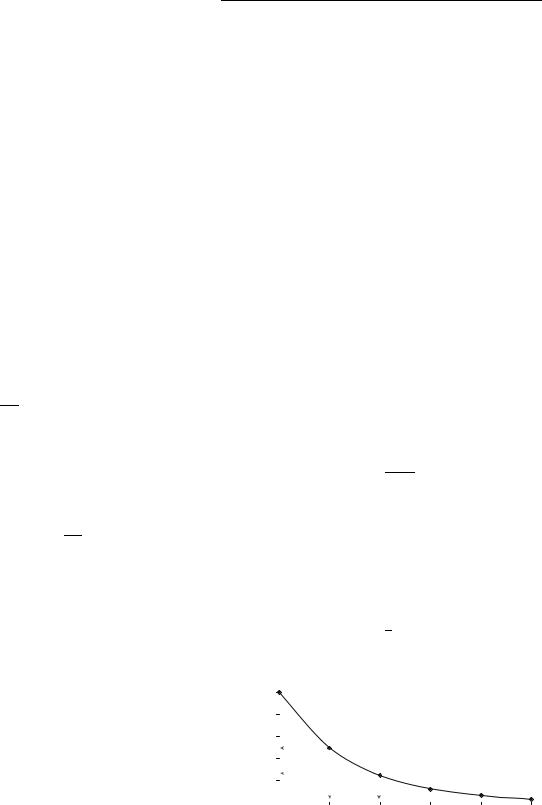
RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY |
561 |
annihilation photons) and radiotherapy (b /þ and Auger electrons) applications.
64Cu |
33:4 h |
64Zn |
|
0 b |
|
|
|
|
|
|
g |
|
Q |
|
|
||
þ |
þ |
|
n |
e þ |
þ |
|
|
||||||||||
29 |
! |
30 |
1 |
|
|
|
|
|
|
|
|||||||
64Cu |
66:8 h |
64Ni |
þ |
0 bþ |
þ |
n |
e þ |
|
Q |
þ |
2 |
|
0:511 MeV photons |
||||
29 |
! |
28 |
þ1 |
|
|
|
|
|
|
|
|||||||
64Cu |
þ |
e 30:2 h |
64Ni |
þ |
n |
þ |
Q |
þ |
Auger e =X rays |
||||||||
29 |
! |
28 |
|
e |
|
|
|
|
|
||||||||
All of the above modes of radioactive decay have been utilized or are under investigation for utilization in the development of radiopharmaceuticals for nuclear medicine applications (either diagnostic imaging or radiotherapy).
RATES OF RADIOACTIVE DECAY
Radioactive decay is a random process where the number of nuclei in an isotopically pure sample that decay during a given time period is proportional to the number of nuclei (N) present in the sample. If radioactivity is defined as a measure of the rate at which the radioactive nuclei disintegrate, then the number of disintegrations per unit time (dN/dt) from a sample is given by the following expression where l is the decay constant (the probability of decay per unit time) and negative sign indicates the loss of the radionuclide through the disintegration. This results in a radioactive decay process that follows a first-order rate law:
dN dt
From this definition, the number of disintegrations per unit time (dN/dt) is also known as the activity. For an isotopically pure sample, the number of nuclei N can be calculated knowing Avogadro’s number (NA), the mass number of the isotope (A) in daltons or grams per mole and the sample’s weight (wt) in grams:
Activity ¼ lN ¼ l NAA wt
As with all measurements, several systems of units can be used to define the amount of radioactivity in a particular sample. And, as with many measuring systems, there are both ‘‘traditional’’ units and new SI units (International System of Units). The original units are referenced to the curie (Ci), named in honor of Madam Curie. A curie is 3.7 1010 (37 billion) radioactive disintegrations per second, or approximately the amount of radioactivity in a gram of radium, one of the natural radioactive elements with which Madam Curie did her research. For many applications of radiation in medicine or industry, the curie is a relatively large quantity, and so the units of mCi (1/1000th) and microcurie (mCi) (1/1,000,000th) are utilized. On the other hand, a nuclear reactor contains millions of curies of radioactivity and units of kilocurie (kCi) (1000) and megacurie (MCi) (1,000,000) are sometimes used. Although the unit of the curie is being supplemented with the newer SI unit, it is still very much in common use.
The SI unit of radioactivity, the becquerel (Bq), is named after Henri Becquerel, the discoverer of radioactivity. It is defined as one disintegration per second. Thus it is much smaller than the Ci. Multiples of becquerel
Table 1. Units of Activity
|
Becquerels or |
Becquerels or |
|
|
|
Curies |
disintegrations s 1 |
disintegrations s 1 |
Curies |
||
1 MCi |
3.7 |
1016 |
1 Bq |
2.7 10 11 |
|
1 kCi |
3.7 |
1013 |
1 kBq |
2.7 |
10 8 |
|
|
10 |
|
|
5 |
1 Ci |
3.7 |
10 7 |
1 MBq |
2.7 |
10 |
1 mCi |
3.7 104 |
1 GBq |
0.027 |
||
1 mCi |
3.7 10 |
|
|
|
|
are the kilobecquerel (kBq) (1000), megabecquerels (MBq) (1,000,000), and so on. These units are summarized in Table 1.
The rate at which radioactive decay occurs can be defined as the half-life, or the amount of time that it takes for one-half of the radiation to decay. Unfortunately, two half-lives do not eliminate it (i.e., one-half decaying in one half-life and then the second one-half decaying in a second half-life), but rather the half-life is always referring to how much is left at any given point in time. Thus, one half-life reduces the radioactivity to one-half or 50%; two half-lives to one-half of one-half, one-fourth or 25%; three half-lives to one-eighth or 12.5%; and so on This leads to a kinetic model that is described by an exponential function (see Fig. 1), which is the solution to the previous equation:
NðtÞ ¼ N0e lt
The half-life is related to the physical decay constant (l) by the simple expression:
0:693
t1=2 ¼ l
As a radionuclide decays, the number of nuclei (N) changes, and the amount of radioactivity present at any given time ‘‘t’’ remaining from an initial amount Ao (expressed in either Bq or Ci) can be given by any of the following:
AðtÞ ¼ A0e lt
AðtÞ ¼ A0eð0:693=t1=2Þt
1 t=t1=2
AðtÞ ¼ A0 2
NumberRelativeof NucleiRadioactive |
100 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
80 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
60 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
50% at 1 Half-life |
|
|
|
||||
|
40 |
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
25% at 2 Half-lives |
|
|
||
|
20 |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0 |
1 |
2 |
3 |
4 |
5 |
||||||
Number of Half-Lives
Figure 1. Relative number of radioactive nuclei remaining after various half-lives of decay.

562 RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY
Radioactive nuclei have half-lives that range from fractions of a second to billions of years. The radioactive material introduced into the body for medical purposes would typically have half-lives on the order of a few hours to a few days, so that it decays away to negligible levels in a relatively short amount of time. At the other extreme, some of the naturally radioactive nuclei in our environment have half-lives of billions of years and the reason they are still present is that they have not had enough time to decay to negligible levels since the earth was formed. An example of such a radionuclide is 40K, which makes up 0.0117% of naturally occurring potassium and has a half-life of 1.27 109 years.
Another interesting case of radioactive decay involves parent–daughter relationships in which a radioactive parent decays to a radioactive daughter. This process is utilized extensively in the medical profession to provide a long-lived source for a short-lived radioisotope in what is known as a generator. The daughter can be obtained from the generator by ‘‘milking’’ it, taking advantage of the differences in chemistry between the parent and daughter elements to extract the daughter off of an ion column,
which holds the parent.
The use of 99mTc, which has a 6 h half-life, is a case in point. Within a 24 h period it has decayed through four half-lives and is only one-sixteenth of its original value. Thus, 99mTc would have to be made in a nuclear facility and shipped daily to meet hospital needs. Fortunately, the
daughter 99mTc is produced by the decay of a parent isotope 99Mo, which has a 2.75 day half-life. A supply of 99mTc can
thus be obtained over a period of 1 week from the more slowly decaying 99Mo.
99Mo |
2:75 days |
99mTc |
|
b |
|
|
6 h |
99Tc |
|
g |
þ |
þ |
n |
þ |
|||||||
42 |
! |
43 |
|
|
e ! |
43 |
|
The kinetics of the quantity of a daughter isotope (d) available from a parent isotope (p) can be readily solved using a first-order differential equation resulting in a straightforward algebraic expression:
Adaughter |
¼ |
ld |
1 eðld lpÞt |
Aparent |
ld lp |
||
|
|
|
|
For most practical cases where the parent is longer lived than the daughter, the daughter activity reaches a value close to the activity of the parent after approximately four half-lives. Thus, in an undisturbed generator that has been allowed to reach equilibrium, the quantity of daughter present (in Ci or Bq) is approximately equal to the activity of the parent at that time. Once the daughter has been extracted (or milked), it immediately begins building up again to a new equilibrium value as shown in Fig. 2.
Also of importance is the time it takes for the daughter to reach its maximum value, which is determined by the decay constants of the two isotopes involved:
lnðldÞ
tmax ¼ lp
ld lp
Again assuming a typical case where the parent is longer lived than the daughter, this is largely deter-
|
1 |
|
|
|
|
|
|
|
0.9 |
|
|
|
|
|
|
Activity |
0.8 |
|
|
|
|
|
|
0.7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0.6 |
|
|
|
|
|
Mo-99 |
Relative |
0.5 |
|
|
|
|
|
|
|
|
|
|
|
Tc-99m |
||
0.3 |
|
|
|
|
|
||
|
0.4 |
|
|
|
|
|
|
|
0.2 |
|
|
|
|
|
|
|
0.1 |
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
0 |
10 |
20 |
30 |
40 |
50 |
60 |
|
|
|
|
Time (hours) |
|
|
|
Figure 2. Shows build-up of 99mTc activity from 99Mo initially and following elution.
mined by the half-life of the daughter. Thus, the daughter will reach 50% of the equilibrium value in one half-live, 75% of the equilibrium value in two halvelives, and so on. The time between subsequent extractions of a daughter from a generator is set by this regeneration time. For the 99Mo ! 99mTc system, the regeneration time needed to reach the maximum amount of daughter 99mTc is very close to 24 h, a very convenient amount of time for routine hospital procedures and is just a fortuitous consequence of the halflives of the two isotopes involved.
Other examples of parent–daughter generators that
are currently used or are under development for future use are 188W/188Re generators, 90Sr/90Y generators,
82Sr/82Rb direct infusion generators, 62Zn/62Cu generators, 224Ra/212Bi or 224Ra/212Pb generators, which also are a source of 212Bi, the daughter product of 212Pb, 225Ac/213Bi
generators and 68Ge/68Ga generators. (For more information on generators see Ref. 5 and references cited therein.)
Specifically,
82Sr |
|
25:4 days |
82Rb |
|
1:6 min |
82Kr |
þ |
bþ |
||
38 |
! |
37 |
! |
36 |
|
|
||||
68Ge |
271 days |
68Ga |
68 min |
68Zn |
þ |
bþ |
||||
32 |
|
! |
31 |
|
! 30 |
|
|
|||
are two generators that can produce short lived PET isotopes, with 82Rb being used for PET studies of myocardial function (rubidium acts as a potassium ion mimic).
The following three are examples of generators for beta or alpha emitting radioisotopes as possible therapeutic agents:
188W |
69:4 days |
188Re |
|
17 h |
188Zn |
þ |
b |
|||
74 |
! |
75 |
! |
|
76 |
|
|
|||
166Dy |
3:4 days |
166Ho |
26:8 h |
|
166 Er |
þ |
b |
|||
66 |
! |
67 |
|
! |
68 |
|
|
|||
212 |
10:64 h |
212 |
|
60:6 min |
208 |
|
|
|
|
|
82Pb |
! |
83Bi |
! |
|
81Tl þ a |
|
||||
RADIONUCLIDE PRODUCTION
The radionuclides used in medicine and the life sciences are produced through neutron and charged particle
RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY |
563 |
induced nuclear reactions. As with chemical reactions, the yield of the product (radioisotope) of interest will depend on the nuclear reaction employed, the energetics of the reaction, the probability of competing reaction pathways, and the ability to separate the desired product from the reactants (target) and any additional nuclear reaction products. For a complete overview of the practice and theory of nuclear reactions, the interested reader is referred to an introductory text on nuclear and radiochemistry (6–9).
Proton-rich radionuclides that decay by positron emission and/or electron capture are produced at cyclotrons and accelerators through charged particle induced nuclear reactions. For example, the production of the commonly used PET radionuclide 18F through the bombardment of a target of 18O-enriched water with high energy protons is produced by the following nuclear reaction:
11p þ 188 O ! 189 F þ 10n þ Q
that is written in a short-hand notation as 18O(p,n)18F. Note that, as in radioactive decay, charge (number of protons), mass number, and total energy are conserved in the nuclear reaction. The Q value for the reaction is the difference in energy (mass) between the reactants and products and is readily calculated from the measured mass excess (D) values for the reactants and products
XX
Q ¼ Dreactants Dproducts
If Q is < 0, then the reaction is endoergic and energy must be supplied to the reaction (through the kinetic energy of the projectile) and if Q is >0, the reaction is exoergic and energy is released in the nuclear reaction. For the 18O(p,n)18F reaction,
Q¼ D11p þ D188O ðD10n þ D189 FÞ
¼0:782 þ 7:289 ð8:071 þ 0:873Þ
¼2:44 MeV
and the proton must supply 2.44 MeV of energy to the reaction. In practice, the actual proton bombarding energy is higher than the Q value because (1) not all of the kinetic energy of the proton is available for the nuclear reaction because of momentum conservation in the collision and (2) the probability of the reaction is typically very low at the threshold energy (Q). Typical conditions for the 18O(p,n)18F reaction on a water target with an in-house cyclotron are 16.5 MeV at 100 mA yielding 3–4 Ci/h. Even in those cases when the Q value for the reaction is >0, the incoming charged particle must have sufficient kinetic energy to overcome the coulomb or charge barrier between the projectile and the target nucleus and any angular momentum barrier that might exist for the reaction.
The probability that the projectile will strike the target nucleus and produce the radioisotope of interest is quantified with the reaction cross-section (s) that has the dimensions of area. While quite sophisticated models exist for predicting reaction cross-sections based upon the underlying nuclear physics, the simple physical
analogy for the cross-section is the area that the target nucleus presents to the incoming beam of projectiles. The
SI unit for cross-section is m2, but the more common unit is a barn (b); one barn is equal to 10 24 cm2 (or 10 28 m2).
The magnitude of a barn can be understood from the fact that a target nucleus with mass number 100 has a radius on the order of 6.5 10 15 m and a ‘‘cross-sectional area’’ of 1.3 10 28 m2 or 1.3 b. In the simplest case when a charged particle beam is bombarding a target that is ‘‘thin’’ enough so that the beam does not lose any appreciable energy in passing through the target, then the production rate (R) for the radioisotope of interest is equal to
R ¼ n I t s
where n is the target nuclei density (nuclei cm 3), I is the number of incident particles per unit time (particles s 1), t is the target thickness (cm), and s is the reaction crosssection (cm2). In most cases, radioisotope production is performed using a thick target and an estimate of the reaction production rate takes into account the variation in the reaction cross-section with projectile energy, since the charged-particle beam loses energy as it passes through or stops in the thick target. Charged particle induced reactions used to produce medical radioisotopes have maximum cross-sections on the order of millibarns.
Neutron-rich radioisotopes that decay by negatron or b emission are produced primarily through neutron-induced nuclear reactions at nuclear reactors. The most commonly used reactions are direct production through single or double neutron capture, direct production through neutron induced fission, and indirect production through neutron capture followed by radioactive decay. An example of direct neutron capture is the production of the 166Ho through the irradiation of 165Ho in a nuclear reactor by the following reaction:
10n þ 16567Ho ! 16667Ho þ Q
Like all neutron capture reactions used to produce medical radioisotopes, this reaction is exoergic with a reaction Q value 6.24 MeV. In contrast to charged-particle induced reactions, there is no coulomb or charge barrier for neutron induced reactions and the cross-section for the neutron capture reaction increases as the energy of the neutron decreases. This increase in cross-section can be understood in that the wavelength of the neutron, and hence its probability of interacting with the target nucleus, increases as the kinetic energy or velocity of the neutron decreases. The maximum yield for most neutron capture reactions is obtained by irradiating the target material in a region of the nuclear reactor where the high energy neutrons produced by fission have been slowed down or moderated so that they have an average kinetic energy of 0.025 eV. One advantage of producing radioisotopes through neutron capture reactions is that the cross-sections are, at 0.025 eV, on the order of barns, whereas charged particle induced cross-sections have peak values on the order of millibarns. A second advantage is that many different targets can be irradiated at the same time in a nuclear reactor, while most accelerator
564 RADIONUCLIDE PRODUCTION AND RADIOACTIVE DECAY
facilities can only irradiate one or two targets at a time. The obvious disadvantage of neutron induced reactions is that the isotope production must be performed off-site at a nuclear reactor, whereas compact cyclotrons can be sited at or near the medical facility making it possible to work with quite short-lived proton-rich isotopes.
The production rate (R) of a radionuclide during irradiation with the moderated or ‘‘thermal’’ neutrons in a nuclear reactor is given by
R ¼ N F s
where N is the number of nuclei of the target isotopes in the sample, F is the flux of thermal neutrons (n cm 2 s 1), and s is the thermal neutron reaction cross-section (cm2). Because those radionuclides created during irradiation can decay during the production process, the activity (A) in Bq of a radionuclide with decay constant l produced by irradiating a sample either in a reactor or with a charged particle beam is given by
A ¼ Rð1 e l ti Þe l td
where R is the production rate for the reaction, ti is the irradiation time, and td is the amount of time the sample
has been allowed to decay following the irradiation. Using again the example of 165Ho(n,g) 166Ho reaction,166Ho has a
half-life of 1.12 days, 165Ho has a thermal cross-section of
58 b and an isotopic abundance of 100%. Irradiation of 87 mg of 165Ho [100 mg target of holmium oxide (Ho2O3)] for 5 days in a thermal neutron flux of 1 1014 n cm 2 s 1 will produce 1.8 1012 Bq or 48 Ci of 166Ho with a specific activity of 0.55 Ci of 166Ho mg 1 of Ho in the target. The
specific activity that can be achieved in the overall (nuclear and chemical) production process is a critical consideration when the radionuclide is used for therapy because it represents the fraction of the atoms in the sample that will have radio-therapeutic activity. In the example of 166Ho, a specific activity of 0.55 Ci mg 1 means that only 1 out of every 1300 Ho atoms in the sample that will be incorporated into the therapeutic agent are radioactive. A disadvantage of using direct neutron capture reactions is that the specific activity of the product radioisotope cannot be improved through chemical means (the product is an isotope of the target).
The parent material of the most commonly used medical radionuclide (99mTc) is produced in high specific activity through the neutron induced fission of 235U. On average, every 100 thermal neutron induced fissions of 235U produce six 99Mo atoms. Because the molybdenum produced in the uranium target can be chemically separated from the uranium and other fission products, and because all of the other molybdenum isotopes have much shorter halflives than 99Mo, the process results in a sample in which nearly every molybdenum atom is 99Mo. While there are a number of advantages from using this ‘‘carrier free’’ (i.e., 100% of the Mo is radioactive 99Mo) 99Mo to create the commercial 99Mo/99mTc generators, the process does create significant amounts of waste that must be appropriately disposed. Neutron-rich radionuclides can also be produced in high specific activity through an indirect method that utilizes neutron capture followed by radioactive decay.
Consider for example the production of 105Rh from the irradiation of ruthenium target. Neutron capture on 104Ru produces 105Ru through the following reaction:
10n þ 10444Ru ! 10544Ru þ Q
105Ru is radioactive (t1/2 ¼ 4.44 h) and beta decays into 105Rh, which has a half-life of 35.4 h. As in the fission case,
the rhodium in the target can be chemically separated from the ruthenium target to produce a sample that is essentially carrier free.
SUMMARY
The use of radionuclides in medicine has been, and continues to be, important to diagnosis of disease and radiotherapy. Many diagnostic agents based on 99mTc are part of the arsenal of radiopharmaceuticals available to the physician. The last 10 years has seen the FDA approval of three new radiotherapeutic agents, namely, Quadramet (a bone pain palliation agent containing 153Sm) and the first two FDA approved radioimmunoconjugates, Zevalin [a 90Y labeled monoclonal antibody that specifically targets the CD20 antigen expressed on >90% of non-Hodgkin’s lymphomas (NHL), and Bexxar (an 131I labeled monoclonal antibody that specifically targets the CD20 antigen expressed on >90% of non-Hodgkin’s lymphomas]. The development of new radiodiagnostic and radiotherapeutic agents will continue and will undoubtedly take advantage of the advances occurring in molecular biology and genomics. Radioactive decay and radionuclide production are two important aspects in the design of new radiopharmaceuticals. For more detailed discussions on these topics, the reader is referred to general textbooks on nuclear and radiochemistry (6–8).
BIBLIOGRAPHY
1.Steigman J, Eckelman WC. Nuclear Science Series (NAS- NS-3204). Nuclear Medicine, The Chemistry of Technetium in Medicine. National Academy Press; Washington (D.C.): 1992.
2.Jurisson SS, Lydon JD. Potential Technetium Small Molecule Radiopharmaceuticals. Chem Rev 1999;99:2205–2218.
3.Brucer M. In: Sorenson JA. et al., editors. The Heritage of Nuclear Medicine. The Society of Nuclear Medicine; New York: 1979.
4.Science 1946;103(2685): 698–705.
5.Lever SZ, Lydon JD, Cutler CS, Jurisson SS. Radioactive Metals in Imaging and Therapy. In: Meyer T, McCleverty J, editors. Comprehensive Coordination Chemistry II Volume 9. London: Elsevier Ltd.; 2004. pp 883–911.
6.Friedlander G, Kennedy JW, Macias ES, Miller JM. Nuclear and Radiochemistry. 3rd ed. New York: Wiley; 1981.
7.Choppin G, Rydberg J, Liljenzin JO. Radiochemistry and Nuclear Chemistry. 2nd ed. Oxford: Butterworth-Heinemann Ltd; 1995.
8.Ehmann WD, Vance DE. Radiochemistry and Nuclear Methods of Analysis. New York: Wiley; 1991.
9.Loveland WD, Morrissey D, Seaborg GT. Modern Nuclear Chemistry. New York: Wiley; 2005.
See also NEUTRON ACTIVATION ANALYSIS; TRACER KINETICS.