

Therapeutic Micro-Nano Technology BioMEMs - Tejlal Desai & Sangeeta Bhatia
.pdf234 |
SAMIR MITRAGOTRI |
[32]M.E. Johnson, S. Mitragotri, A. Patel, D. Blankschtein, and R. Langer. Synergistic effect of ultrasound and chemical enhancers on transdermal drug delivery. J. Pharm. Sci., 85:670–679, 1996.
[33]J.A. Kleinkort and F. Wood. Phonophoresis with 1 percent versus 10 percent hydrocortisone. Phys. Ther., 55:1320–1324, 1975.
[34]J. Kost and R. Langer, Ultrasound-mediated transdermal drug delivery. In V.P. Shah and H.I. Maibach (eds.)
Topical Drug Bioavailability, Bioequivalence, and Penetration. Plennum: New York. pp. 91–103, 1993,
[35]J. Kost, U. Pliquett, S. Mitragotri, A. Yamamoto, J.Weaver, and R. Langer, Enhanced transdermal delivery: synergistic effect of ultrasound and electroporation. Pharm. Res., 13:633–638, 1996.
[36]J. Kost, S. Mitragotri, and R. Langer. Phonophoresis. In R. Bronaugh and H.I. Maibach (eds.) Percutaneous Absorption,. pp. 615–631, 1999.
[37]J. Kost, S. Mitragotri, R. Gabbay, M. Pishko, and L. R. Transdermal extraction of glucose and other analytes using ultrasound. Nat. Med., 6:347–350, 2000.
[38]L. Le, J.Kost, and S. Mitragotri. Combined effect of low-frequency ultrasound and iontophoresis: applications for transdermal heparin delivery. Pharm. Res., 17:1151–1154, 2000.
[39]M. Machluf and J. Kost. Ultrasonically enhanced transdermal drug delivery. Experimental approaches to elucidate the mechanism. J. Biomat. Sci., 5:147–156, 1993.
[40]G. Menon, D. Bommanon, and P. Elias. High-frequency sonophoresis: permeation pathways and structural basis for enhanced permeability. Skin Pharmacol., 7:130–139, 1994.
[41]S. Mitragotri, D. Edwards, D. Blankschtein, and R. Langer. A mechanistic study of ultrasonically enhanced transdermal drug delivery. J. Pharm. Sci., 84:697–706, 1995.
[42]S. Mitragotri, D. Blankschtein, and R. Langer. Sonophoresis: ultrasound mediated transdermal drug delivery. In J. Swarbrick, Boylan, J. (eds.) Encl. of Pharm. Tech., Marcel Dekker, 1995.
[43]S. Mitragotri, D. Blankschtein, and R. Langer. Transdermal drug delivery using low-frequency sonophoresis. Pharm. Res., 13:411–420, 1996.
[44]S. Mitragotri, D. Blankschtein, and R. Langer. Sonophoresis: enhanced transdermal drug delivery by application of ultrasound. In S. J. and J. Boylan, (eds.), Encyl. Pharm. Tech.,. pp. 103–122, 1996.
[45]S. Mitragotri, D. Blankschtein, and R. Langer. An explanation for the variation of the sonophoretic transdermal transport enhancement from drug to drug. J. Pharm. Sci., 86:1190–1192, 1997.
[46]S. Mitragotri, J. Farrell, T. H., T. Terahara, J. Kost, and R. Langer. Determination of the threshold energy dose for ultrasound-induced transdermal drug delivery. J. Control. Rel., 63:41–52, 2000.
[47]S. Mitragotri, D. Ray, J. Farrell, H. Tang, B. Yu, J. Kost, D. Blankschtein, and R. Langer. Synergistic effect of ultrasound and sodium lauryl sulfate on transdermal drug delivery. J. Pharm. Sci., 89:892–900, 2000.
[48]S. Mitragotri and J. Kost. Transdermal delivery of heparin and low-molecular weight heparin using lowfrequency ultrasound. Pharm. Res., 18:1151–1156, 2000.
[49]S. Mitragotri and J. Kost. Low-frequency sonophoresis: a non-invasive method for drug delivery and diagnostics. Biotech. Progress, 16:488–492, 2000.
[50]S. Mitragotri. Synergistic effect of enhancers for transdermal drug delivery. Pharm. Res., 17:1354–1359, 2000.
[51]W.S. Quillen. Phonophoresis: a review of the literature and technique. Athelet. Train., 15:109–110, 1980.
[52]A. Tezel, A. Sanders, J. Tuchscherer, and S. Mitragotri. Synergistic effect of low-frequency ultrasound and surfactant on skin permeability. J. Pharm. Sci., 91:91–100, 2001.
[53]T. Terahara, S. Mitragotri, and R. Langer. Porous resins as a cavitation enhancer for low-frequency sonophoresis. J. Pharm. Sci., 91:753–759, 2002.
[54]T. Terahara, S. Mitragotri, J.Kost, and R. Langer. Dependence of low-frequency sonophoresis on ultrasound parameters; distance of the horn and intensity. Int. J. Pharm., 235:35–42, 2002.
[55]H. Tang, D. Blankschtein, and R. Langer. Effects of low-frequency ultrasound on the transdermal penetration of mannitol: comparative studies with in vivo and in vitro studies. J. Pharm. Sci., 91:1776–1794, 2002.
[56]H. Tang, S. Mitragotri, D. Blankschtein, and R. Langer. Theoretical description of transdermal transport of hydrophilic permeants: application to low-frequency sonophoresis. J. Pharm. Sci., 90:543–566, 2001.
[57]A. Tezel, A. Sens, and S. Mitragotri. Investigations of the role of cavitation in low-frequency sonophoresis using acoustic spectroscopy. J. Pharm. Sci., 91:444–453, 2002.
[58]A. Tezel, A. Sens, and S. Mitragotri. A theoretical analysis of low-frequency sonophoresis: dependence of transdermal transport pathways on frequency and energy density. Pharm. Res., 19:1841–1846, 2002.
[59]A. Tezel, A. Sens, and S. Mitragotri. A theoretical description of transdermal transport of hydrophilic solutes induced by low-frequency sonophoresis. J. Pharm. Sci., 92:381–393, 2003.
TRANSDERMAL DRUG DELIVERY USING LOW-FREQUENCY SONOPHORESIS |
235 |
[60]K. Tachibana. Transdermal delivery of insulin to alloxan-diabetc rabits by ultrasound exposure. Pharm. Res., 9:952–954, 1992.
[61]K. Tachibana and S. Tachibana. Use of ultrasound to enhance the local anesthetic effect of topically applied aqueous lidocaine. Anestheiology, 78:1091–1096, 1993.
[62]K. Tachibana and S. Tachibana. Transdermal delivery of insulin by ultrasonic vibration. J. Pharm. Pharmacol., 43:270–271, 1991.
[63]A.R.Williams. Phonophoresis: an in vivo evaluation using three topical anaesthetic preparations. Ultrasonics, 28:137–141, 1990.
[64]R. Alvarez-Roman, G. Merriono, Y.N. Kalia, A. Naik, and R. Guy. Skin permeability enhancement by low-frequency sonophoresis-lipid extraction and transport pathways. J. Pharm. Sci., 92:1138–1146, 2003.
[65]G. Merrino, Y.N. Kalia, and R.H. Guy. Ultrasound-enhanced transdermal transport. J. Pharm. Sci., 92:1125– 1137, 2003.
[66]G. Merriono, Y.N. Kalia, M.B. Delgado-Charro, R. Potts, and R.H. Guy. Frequency and thermal effects on the enhancement of transdermal transport by sonophoresis. J. Control. Rel., 88:85–94, 2003.
[67]L.J. Weimann and J. Wu. Transdermal delivery of poly-l-lysine by sonomacroporation. Ultrasound Med. Biol., 28:1173–1180, 2002.
[68]A. Joshi and J. Raje. Sonicated transdermal drug transport. J. Control. Rel., 83:13–22, 2002.
[69]L. Machet and A. Boucaud. Phonophoresis: efficiency, mechanisms, and skin tolerance. Int. J. Pharm., 243:1–15, 2002.
[70]A. Boucaud, M.A. Garrigue, L. Machet, L. Vaillant, and F. Patat. Effect of sonication parameters on transdemral delivery of insulin to hairless rats. J. Pharm. Sci., 91:113–119, 2002.
[71]K. Fellinger and J. Schmidt. Klinik and Therapies des Chromischen Gelenkreumatismus. Maudrich Vienna, Austria, pp. 549–552, 1954.
[72]G.L. Coodley. Bursitis and post-traumatic lesions. Am. Pract., 11:181–187, 1960.
[73]J.E. Griffin. Physiological effects of ultrasonica energy as it is used clinically. J. Am. Phys. Ther. Assoc., 46:18–26, 1966.
[74]J.C. McElnay, M.P. Matthews, R. Harland, and D.F. McCafferty. The effect of ultrasound on the percutaneous absorption of lingocaine. Br. J. Clin. Pharmacol., 20:421–424, 1985.
[75]M.A. Moll. New approaches to pain. US Armed Forces Med. Serv. DIg., 30:8–11, 1979.
[76]D. Hofman and F. Moll. The effect of ultrasound on in vitro liberation and in vivo penetration of benzyl nicotinate. J. Control. Rel., 27:187–192, 1993.
[77]D. Levy, J. Kost, Y. Meshulam, and R. Langer. Effect of ultrasound on transdermal drug delivery to rats and guinea pigs. J. Clin. Invest., 83:2974–2078, 1989.
[78]S. Mitragotri and J. Kost. Low-frequency sonophoresis: a review. Adv. Drug Deliv. Rev., 56:589–601, 2004.
[79]C.M. Gockel, S. Bao, and K.W. Beagley. Transcutaneous immunization induces mucosal and systemic immunity:a potent method for targeting immunity to the female reproductive tract. Mol. Immunol., 37:37– 544, 2000.
[80]P.L. Ogra, J. Mestecky, M.E. Lamm, W. Strober, J.R. McGhee, and J. Bienenstock. Handbook of Mucosal Immunology. New York: Acedemic Press, 1994.
[81]T. Scharton-Kersten, J.Yu, R.Vassell, D. O’Hagan, C.R. Alving, and G.M. Glenn. Transcutaneous immunization with bacterial ADP-ribosylating exotoxins, subunits, and unrelated adjuvants. Infect. Immun., 68:5306–5313, 2000.
[82]I. Roitt, J. Brostoff, and D. Male. Immunology,(5th Ed.). London: Mosby, 1998.
[83]Tana, S.Watarai, E. Isogai, and K. Oguma. Induction of intestinal IgA and IgG antibodies preventing adhesion of verotoxin-producing Escherichia coli to Caco-2 cells by oral immunization with liposomes.
Lett. Appl. Microbiol., 36:135–139, 2003.
[84]S. Mitragotri, M. Coleman, J. Kost, and R. Langer. Transdermal extraction of analyets using low-frequency ultrasound. Pharm. Res., 17:466–470, 2000.
[85]S. Mitragotri, M. Coleman, J. Kost, and R. Langer. Analysis of ultrasonically extracted interstitial fluid as a predictor of blood glucose values. J. Appl. Physiol., 89:961–966, 2000.
[86]J. Kost, S. Mitragotri, T. Elstrom, N. Warner, and S. Kellogg. Ultrasonic Skin Permeabilizing Device for Transdermal Glucose Monitoring. 2nd La Jolla Conference on Diabetes, San Diego, 2003.
[87]A.J. Singer, C.S. Homan, A.L. Church, and S.A. McClain. Low-frequency sonophoresis: pathologic and thermal effects in dogs. Acad. Emerg. Med., 5:35–40, 1998.
236 |
SAMIR MITRAGOTRI |
[88]A. Boucaud, J. Montharu, L. Machet, B. Arbeille, M.C. Machet, F. Patat, and L. Vaillant. Clinical, histologic, and electron microscopy study of skin exposed to low-frequency ultrasound. Anatom. Rec., 264:114–119, 2001
[89]N.P. Katz, D.E. Shapiro, T.E. Hermann, J. Kost, and L. Custer. Rapid onset of cutaneous anesthesia with EMLA cream after pretreatment with a new ultrasound emitting device. Anesth. Analg., 98:371–376, 2004.
[90]Simmonin, On the mechanisms of in vitro and in vivo phonophoresis. J. Control. Rel., 33:125–141, 1995.
[91]A. Tezel and S. Mitragotri. Interactions of inertial cavitation collapses with stratum corneum lipid bilayers. Biophys. J. (in press), 2003.
[92]K.S. Suslick. Ultrasound:Its Chemical, Physical and Biological Effects. VCH Publishers, 1989.
[93]A. Tezel and S. Mitragotri. Interactions of inertial cavitation bubbles with stratum corneum lipid bilayers during low-frequency sonophoresis. Biophys. J., 85:3502–3512, 2003.
[94]J. Blake and D. Gibson. Cavitation bubbles near boundaries. In J. Lumley, M. Van Dyke and H. Reed (eds.),
Annual Reviews of Fluid Mechanics, pp. 99–123, 1985.
[95]Naude and A. Ellis. On the mechanisms of cavitation damage by non-hemispherical cavities in contact with solid boundary. Trans. ASME J. Basic Eng., 83:648–556, 1961.
[96]T. Benjamin and A. Ellis. The collapse of cavitation bubbles and the pressures thereby produced against solid boundaries. Philos. Trans. R. Soc. London Ser A, 260:221–240, 1966.
[97]W. Lauterborn and H. Bolle. Experimental investigations of cavitation bubble collapse in the neighbourhood of a solid boundary. J. Fluid. Mech., 72:391–399, 1975.
[98]M. Plesset and R. Chapman. Collapse of an initially spherical vapour cavity in the neighbourhood of a solid boundary. J. Fluid. Mech., 47:283–290, 1971.
[99]S. Mitragotri. Effect of bilayer disruption on transdermal transport of low-molecularweight hydrophobic solutes. Pharm. Res., 18:1022–1028, 2001.
[100]G. Black. Interaction between anionic surfactants and skin. In K.Walters and J. Hadgraft, (eds.), Pharmaceutical Skin Penetration Enhancement, Marcel Dekker, New York, Basel, Hong Kong, pp. 145–174, 1993.
[101]K.A. Walters, Surfactants and percutaneous absorption. In R.C. Scott, R.H. Guy, and J. Hadgraft (eds.), Predictions of Percutaneous Penetration, IBC Technical Services: London. pp. 148–162, 1990.
14
Microdevices for Oral
Drug Delivery
Sarah L. Tao1 and Tejal A. Desai2
1 Department of Biomedical Engineering, Boston University Boston, MA 02215
2 Department of Bioengineering and Physiology, University of California, San Francisco, CA
14.1. INTRODUCTION
The application of microand nanotechnology to the biomedical arena has tremendous potential in terms of developing new diagnostic and therapeutic modalities. Recent advances in micromachining and microelectriomechanical systems (MEMS) technology have provided the opportunity to fabricate miniature biomedical devices for a variety of applications. While the majority of research has focused on the development of miniaturized diagnostic tools, researchers have more recently concentrated on the development of microdevices for therapeutic applications. The progression of microfabrication technology has enabled the creation of entirely new classes of drug delivery devices which can possess a combination of structural, mechanical, chemical, and electronic features to surmount the challenges associated with conventional systems. Some therapeutic applications of microtechnology include microneedles for transdermal delivery [1, 2], as well as various implantable systems, such as immunoisolating biocapules [3, 4] and microchips for controlled release [5, 6]. However in this research, the strengths of microfabrication and micromachining were capitalized to create a completely novel microdevice from silicon dioxide, porous silicon, and poly(methyl methacrylate) for cytoadhesive oral drug delivery with inherent features providing controlled release.
14.1.1. Current Challenges in Drug Delivery
The ultimate aim of pharmacy and medicine is the delivery of any drug at the right time in a safe and reproducible manner to a specific target at the required level [7]. Conventional
238 |
SARAH L. TAO AND TEJAL A. DESAI |
dosage forms, such as oral delivery and injection, are the predominant routes for drug administration. However, these types of dosages are not easily able to control the rate of drug delivery or the target area of the drug and are associated with an immediate, rapid release of drug. Consequently, the initial concentration of the drug in the body peaks above the level of toxicity and then gradually diminishes over time to an ineffective level. The duration of therapeutic efficacy then becomes dependent on the frequency of administration and the halflife of the drug. High dosages of non-targeted drugs are often administered in order to achieve an effective blood concentration. In recent years, increasingly sophisticated and potent drugs have been developed by the biotech industry. For many of these new protein-based and DNA-based compounds, the therapeutic concentration range is small and varies with time, and toxicity is observed for concentration spikes, which renders traditional methods of drug delivery ineffective [8]. An immense amount of interest has been increasingly placed on controlled release drug delivery systems to maintain the therapeutic efficacy of these drugs. There are a number of mechanisms that can provide such controlled release of drugs, including transdermal patches, implants, bioadhesive systems, and microencapsulation. In addition, attractive drug delivery technologies such as inhaled and sustained release injectable peptide/protein drugs are currently under intensive study.
14.1.2. Oral Drug Delivery
Oral drug delivery is one of the most preferred methods of drug administration due to its non-invasive nature. However, it is generally not a viable method for peptide and protein delivery. This is, in part, a consequence of the first-pass effect of the liver, but primarily due to the acidic environment of the stomach and the resistance exerted by the intestine. The human gastrointestinal (GI) tract by nature resists absorption of peptides, proteins, and other macromolecules until they are broken down into smaller molecules. The acidic environment of the stomach combined with an array of enzymes and physical barriers in the intestines either destroy or prevent absorption of nearly all macromolecules. This problem leads to unacceptably low oral bioavailability of the drug, which then fails to induce a clinical response. Several approaches are currently being developed to enhance the oral delivery of macromolecular drugs. Protective coatings, such as lipids and polymers, have been used to protect peptides during transport through the acidic environment of the stomach and improve transport across the intestinal wall [9, 10]. Bioadhesive agents have been used to induce contact of the peptide to the intestinal wall [11–13]. This allows for local delivery of the peptide to sites in the GI tract generating greater levels of absorption and stability. In addition, the use of permeation enhancers to augment uptake and transport through the intestinal wall, and protease inhibitors to protect peptides from enzymatic degradation is being explored [14, 15]. While all these approaches have been shown to improve upon the oral bioavailability of large molecules, none of these approaches offers a complete solution for adequate and safe oral administration.
14.1.3. Bioadhesion in the Gastrointestinal Tract
Bioadhesive drug delivery systems have been given considerable interest due to their potential for delaying transit and prolonging the residence time at the site of drug action or absorption. Bioadhesion refers to the adherence of molecules (bioadhesives) to biological
MICRODEVICES FOR ORAL DRUG DELIVERY |
239 |
surfaces [16]. “Targeting” refers to strategies of effectively directing molecules to a particular tissue, cell type, or subcellular compartment [17]. Localization of the delivery system at a given target site prone to drug action could increase the drug concentration gradient due to intense contact [11,13].
The gastrointestinal tract is the most important body surface for drug administration due to its massive surface area. Two main targets exist for anchoring a delivery system in the gastrointestinal tract: the mucus gel layer and mucosal tissue. Intestinal mucus is composed of high molecular weight glycoproteins. The mucin gel layer covers the mucosal tissue with a continuous adherent blanket. Its thickness varies regionally along the GI tract, decreasing from 50–500 μm in the stomach to 15–150 μm in the colon [11]. Adhesive interactions with the complex structure of mucus gel layer are generally entered through non-specific Van der Waals and/or hydrophobic interactions. The non-specificity of the interaction limits the maximum duration of mucoadhesion to the turnover time of the mucus gel layer. In the intestine, mucus turnover has been estimated to be between 47 and 270 minutes, which is on the same scale as normal intestinal transit (three to four hours) [13, 17, 18]. In this case, drug release and absorption must be completed during this limited adhesion time.
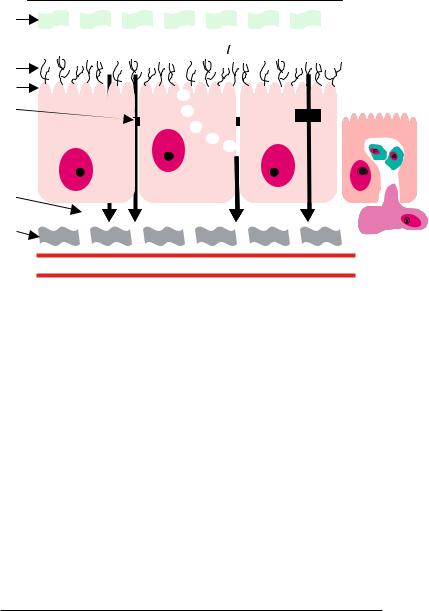
An alternative to mucoadhesion is “cytoadhesion,” where adhesion occurs directly to the cell surface [13]. The mucosal epithelium of the small intestine is comprised of a single layer of polarized epithelial cells, predominantly consisting of enterocytes. The enterocytes are connected through tight intercellular junctions with pore diameters reported to be less than 10 angstroms [17]. Follicle associated epithelium (FAE) is differentiated from other intestinal epithelial sites by the presence of specialized antigen sampling membranous epithelial (M) cells. The transcytotic capacity of these cells is much greater than other intestinal epithelial cell types, and therefore, a potentially important site for drug delivery [19]. Molecules can be transported across the epithelial barrier by passive diffusion through transcellular or paracellular pathways, or actively transported across by membrane-derived vesicles or membrane bound carriers. Uptake can also occur due to adsorptive endocytosis via clathrin-coated pits and vesicles, fluid phase endocytosis, and phagocytosis induced by M cell antigen sampling. Figure 14.1 shows a schematic of the epithelium and the various pathways for molecular transport across the barrier.
Cytoadhesion of a drug delivery system would involve grafting a ligand that shows affinity for a receptor in the gastrointestinal cavity to mediate an adhesive interaction between the particle and the biological surface. Enterocytes and M cells are both potential target sites for cytoadhesion. Although particle adsorption by enterocytes is relatively inefficient when compared to that of M cells, the greater surface area that they occupy offsets their inefficiency.
Lectins, deemed “second generation” bioadhesives, are one group of molecules that are able to recognize surface structures of intestinal cells. Lectins are a class of carbohydrate binding proteins or glycoproteins of non-immune origin. Most lectins contain two or more sugar binding sites and can agglutinate cells and precipitate glycoconjugates with considerable specificity without altering the covalent structure of the glycosyl moiety [11]. Lectins are an ideal possibility for targeted drug delivery because of their relative resistance to low pH and enzymatic degradation. And, because the intestinal epithelial cells maintain a cell surface glycocalyx consisting of membrane-anchored glycoconjugates, a multitude of lectin binding sites exist along the gastrointestinal tract. The structural diversity of cellsurface glycoconjugates encodes unique signals recognized by lectins in a complementary
240 |
SARAH L. TAO AND TEJAL A. DESAI |
Lumen
Mucus Layer
Transcellular |
Paracellular Endo/Transcytosis Carrier Mediated |
Glycocalyx |
|
Microvilli |
|
Tight Junction |
Antigen Sampling |
|
|
Enterocyte |
M cell |
|
|
|
tes |
|
Lymphocytes |
Enterocyte
Interstitium
Macrophage
Basement
Membrane
Capillary
FIGURE 14.1. Schematic of the structure of the epithelium. Molecules can be transported across the epithelial barrier by passive diffusion through transcellular or paracellular pathways, or actively transported across by membrane-derived vesicles or membrane bound carriers. Uptake can also occur due to adsorptive endocytosis via clathrin-coated pits and vesicles, fluid phase endocytosis, and phagocytosis induced by M cell antigen sampling.
way analogous to ligand-receptor interactions [20]. These interactions are even thought to be associated with cell internalization signaling pathways. A number of orally administered plant lectins have been found to bind to the luminal surface of the gut with high affinity (Kd values of 10−4 to 10−6 M) [21]. Their binding is primarily dependent on the structure and accessibility of carbohydrate moieties on the epithelial cells and the sugar specificity of the lectins. Table 14.1 shows examples of commonly found lectins and their sugar binding specificities.
14.1.4. Microdevice Technology
Micromachined platforms, when combined with complementary approaches, may address some of the shortcomings of current oral delivery systems for peptides and proteins by
TABLE 14.1. Examples of common lectins.
Lectin |
Abbreviation |
MW (Da) |
Sugar Specificity |
Triticum vulgaris (Wheat germ) |
WGA |
36,000 |
N-AcGlu;SA |
Lycopersicon esculentum (Tomato) |
TL |
71,000 |
N-AcGlu |
Canavalia ensiformis (concavalin A) |
Con A |
102,000 |
Man;Glu |
Phaseolus vulgaris (Red kidney bean) |
PHA |
115,000 |
Gal;N-AcGal |
Arachis hypogea (Peanut) |
PNA |
120,000 |
β-Gal |
|
|
|
|
MICRODEVICES FOR ORAL DRUG DELIVERY |
241 |
||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
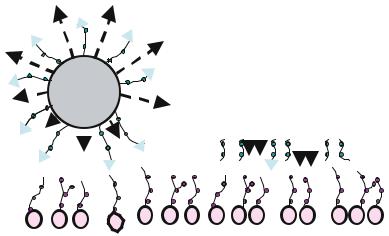
FIGURE 14.2. Microfabricated platforms have many advantages over traditional microsphere delivery systems. In contrast to microspheres, microfabricated devices may be specifically designed flat and thin to maximize contact area with the intestinal lining. This flat design minimizes the side areas exposed to constant flow and can be microfabricated to incorporate single or multiple drug reservoirs to contain a number of drugs/biomolecules of interest. These reservoirs also allow for unidirectional release of the drug. Furthermore, specific areas of the device can be targeted for surface modification.
combining several features into a single drug delivery vehicle [22]. First, one can achieve control over the size and shape of the delivery device [23–25]. Unlike traditional delivery particles, such as microspheres, microfabricated devices may be specifically designed flat, thin, and disc-shaped to maximize contact area with the intestinal lining and minimize the side areas exposed to the constant flow of liquids through the intestines (Figure 14.2). The size of the microdevices can be selected small enough to have good contact with the undulations of the intestinal wall and large enough to avoid endocytosis of the entire particle. Secondly, one can selectively attach bioadhesive agents targeting specific cell in the GI tract onto the device surface using relatively simple surface chemical modification strategies [23,24]. Finally, micromachining presents the opportunity to create multiple reservoirs of a desired size to contain any number of biomolecules of interest which can then be released the shortest distance towards the intestinal epithelium [23–25].
14.2. MATERIALS
The use of silicon as a starting substrate material in the integrated circuit industry was based on its intrinsic mechanical stability and the feasibility of integrating sensing and electronics in self-contained microsystems capable of performing intelligent tasks, such as sensing and signal processing, at a very low manufacturing cost [26, 27]. From its roots in electrical engineering, the discipline of miniaturization has become widely accepted in fields such as mechanical engineering and biomedical engineering. This in turn has led to a wider array of materials used in microfabrication.
242 |
SARAH L. TAO AND TEJAL A. DESAI |
In the realm of microfabrication, even for biomedical applications, silicon remains the starting substrate of choice namely due to the fact that a great deal of equipment has been built to accommodate silicon wafers. As other substrates are harder to accommodate, silicon intrinsically appears to have a convenient advantage. Other advantages of silicon include its superior mechanical stability and well-established chemical techniques for modification utilizing silane chemistry. Although preliminary medical evidence is thought to suggest that silicon remains benign in the body [28], it is not recognized as a Food and Drug Administration (FDA) approved biomaterial. Therefore considerable efforts have been made towards increasing silicon biocompatibility.
14.2.1. Silicon Dioxide
Silicon readily oxidizes to form a stable oxide layer. Silicon oxides usually function as insulation between conducting layers, diffusion and ion implementation masks, capping doped oxides to prevent loss of dopants, for gettering impurities, and for passivation to protect devices from impurities moisture and scratches. In terms of biological applications, this oxide layer may completely isolate underlying layers of a device thereby interacting more favorably with the cellular environment.
14.2.2. Porous Silicon
The formation of porous silicon was first discovered by Uhlir in 1956 [29] and has led to creation of many devices from quantum structures and permeable membranes to photoluminescent and electroluminescent devices. Porous silicon has numerous important
˚
features. Pore size can range in diameter from 20 A to 10 μm and follow crystallographic orientation. It is also highly reactive, and therefore oxidizes and etches at very high rate [27]. Furthermore, recent interest has been gathered in the use of porous silicon in biological applications from biosensors to mammalian cell culture [30–32].
For biomedical applications, polymers have become a favorable alternative to the silicon substrate. Although surface modification protocols for modifying polymers are not as established, polymer substrates still have advantages comparable to silicon. First, various methods can be used to construct the devices inexpensively and in large number, such as injection molding, laser ablation, imprinting, and hot embossing [33]. For this reason, polymers are ideal for the fabrication of disposable devices. Second, high-aspect-ratio microstructure devices can be produced with polymer substrates that are often very difficult to achieve with silicon [33]. Third, polymers can exist either in a hard, glassy state, or a soft rubbery state, an option typically unassociated with structures based on silicon. Fourth, in contrast to silicon, the FDA has already accepted a variety of polymers for internal use. Poly methylmethacrylate (PMMA), polylactic acid (PLA), polyglycolic acid (PGA), and polyethylene (PE) are among such polymers. Devices microfabricated from these polymer alternatives may provide a more suitable interface with biological tissue.
14.2.3. Poly(methyl methacrylate)
PMMA is a synthetic non-biodegradable polymer first discovered in Germany in the early 1900s. In terms of microfabrication, PMMA is used in short wavelength
MICRODEVICES FOR ORAL DRUG DELIVERY |
243 |
photolithographic systems such as deep ultraviolet, electron beam, ion beam, and x-ray lithography. PMMA is a model substrate for microfabricated systems geared towards biomedical application. Besides being used extensively as a resist in photolithographic applications, PMMA is known to be biocompatible and is FDA approved. It is already used in many medical applications such as bone cement [34, 35], contact lenses [36], and intraocular lenses [37].
14.3. MICROFABRICATION
The overall concept of constructing a micromachined platform for drug delivery remains the same across all three substrates (silicon dioxide, porous silicon and PMMA). As a prototype, rectangular-shaped microparticles were fabricated with dimensions that would potentially allow for in vivo transit. The rectangular dimensions were chosen for simplicity of design and fabrication, the rectangle being the simplest shape which photomask generating equipment can create. However, by simply changing the photomask features, any shape or size particle can be easily fabricated. The size of the particles may greatly affect the response generated in the body [38]. Although smaller particles, less than five microns, have shown an increased adherence in the whole gut, they are more likely to induce a localized inflammatory response followed by easy uptake by macrophages [39]. In this case, there is an increased risk that the carrier system might be degraded after internalization, followed by loss of its activity [39, 20]. Particles with a much larger size are taken up less effectively by macrophages, however, carrier systems greater than 200 microns may illicit a foreign body response if retained in the GI tract. For these reasons, the devices were fabricated with several dimensions ranging from lengths and widths of 50 to 200 microns. The thickness of the devices is limited by the processing capability of the microfabrication techniques used. Efforts here were concentrated towards maximizing drug loading volume while also maximizing the surface area of the face to side wall ratio. Microdevices between two and 25 microns thick were fabricated. The fabrication processes for creating such a system in the three substrates remains very different. However, these processes were all built upon standard MEMS fabrication techniques including photolithography, etching, and thin film deposition.
14.3.1. Silicon Dioxide [23]
Silicon dioxide microdevices were fabricated on silicon <111> p-type wafers cleaned by the RCA standard three step procedure. An etch stop layer comprised of thermal silicon dioxide was first formed by wet thermal oxidation. Low-pressure chemical vapor deposition (LPCVD) was then used to deposit a sacrificial layer of polysilicon atop the thermal oxide. Next, a layer of low temperature oxide (LTO) was deposited by LPCVD to form the device layer. The processed wafers were then spin-coated with a positive resist and photolithography was performed by exposing the wafers to ultraviolet light through a photomask defining the shape of the device reservoir. Following photoresist development, the exposed LTO device layer was carved by performing either a wet or dry etching procedure. A wet etch created a rounded reservoir and was performed by timed submersion of the masked wafer in a buffered oxide solution followed by excess photoresist removal. A dry etch created a