


13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.2.Качественный анализ
Другой пример: при обнаружении ионов Cr6+ в виде Cr2O 72− мешает ион
Fe3+, обусловливающий также желтую окраску раствора. Ионы железа переводят в бесцветный фосфатный комплекс [Fe(PO4)2]3–.
Экстракция. Для экстракции подбирают такой органический растворитель, чтобы обнаруживаемый и мешающий ионы оказались в разных фазах. Чаще всего в органический слой переводят обнаруживаемый ион. В качестве экстрагентов применяют толуол, бензол, изоамиловый спирт, тетрахлорметан, хлороформ.
Например, обнаружению Co2+ в виде [Co(SCN)4]2– мешает ион Bi3+, образующий с тиоцианатным ионом окрашенное в желтый цвет соединение [Bi(SCN)4]–. В присутствии ионов Bi3+ комплекс [Co(SCN)4]2– экстрагируют амиловым спиртом, а соединение висмута остается в водном растворе.
Осаждение осуществляют с помощью общих реагентов с образованием гидроксидов, карбонатов, сульфидов, сульфатов, фосфатов. Например, мешающее действие катионов p- и d-элементов при обнаружении s- элементов устраняют, применяя гидроксид магния Мg(OH)2:
2Fe3+ + 3Mg(OH)2 = 2Fe(OH)3 + 3Mg2+
2Cr3+ + 3Mg(OH)2 = 2Cr(OH)3 + 3Mg2+ Pb2+ + Mg(OH)2 = Pb(OH)2 + Mg2+ Zn2+ + Мg(OH)2 = Zn(OH)2 + Mg2+
В виде сульфатов осаждают мешающие ионы бария Ba2+ и стронция Sr2+ при обнаружении ионов кальция Ca2+. Сульфат кальция имеет довольно высокую растворимость, что позволяет провести обнаружение Ca2+ в растворе над осадком после осаждения CaSO4.
13.3. Физико-химические методы анализа
Управляемый технологический процесс невозможен без современных методов контроля, которые дают возможность повысить качество продукции. В настоящее время существуют разнообразные и многочисленные методы анализа, которые делятся на химические, инструментальные (физические и физико-химические) и биологические.
Химические методы анализа основаны на химическом превращении анализируемого вещества в новое соединение, которое обладает характерными свойствами и позволяет установить наличие этого вещества или определить его количество.
Инструментальные методы анализа основаны на изучении физиче-
ских свойств веществ или продуктов их реакции с помощью приборов. Эти методы анализа отличаются высокой чувствительностью и быстротой выполнения. Если химические методы анализа (например, гравиметрический) продолжаются несколько часов, а иногда и суток, то анализ вещества современными инструментальными методами обычно занимает несколько минут.
Химия. Учеб. пособие |
-193- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
Сочетание различных инструментальных методов при анализе веществ приводит к возникновению новых «гибридных» методов. Сочетание хроматографии с фотометрией дает хроматофотометрию, существует хромато- масс-спектроскопия.
Из физико-химических методов анализа важное значение имеют: оп-
тические (эмиссионная фотометрия пламени, атомно-абсорбционный, моле- кулярно-абсорбционный, люминесцентный, рефрактометрический, поляриметрический); электрохимические (потенциометрия, кондуктометрия, вольтамперометрия, амперометрическое титрование); хроматографические и радиометрические методы.
13.3.1. Оптическиеметодыанализа
Эти методы анализа основаны на измерении эффектов взаимодействия электромагнитного излучения оптического диапазона с веществом, сопровождающихся излучением, поглощением или отражением лучистой энергии. К оптическому диапазону относят область электромагнитного излучения с длиной волн от 10 нм до 1 000 мкм. Оптический диапазон подразделяют на ультрафиолетовую (10–400 нм), видимую (400–760 нм) и инфракрасную (760 нм–1000 мкм) области спектра. Деление спектра на ряд областей условен и служит только для примерной оценки границ диапазонов.
Оптические методы анализа можно классифицировать по нескольким признакам. По типу взаимодействия электромагнитных волн с вещест-
вом делятся на методы, основанные на измерении эффектов поляризации
(рефрактометрия, интерферометрия и поляриметрия) и спектральные мето-
ды, основанные на поглощении, рассеянии или испускании электромагнитно-
го излучения (атомный и молекулярный спектральные анализы). Деление на атомный и молекулярный спектральный анализ принципиально, т. к. в методах атомной спектроскопии всегда имеют дело с узкими линейчатыми спектрами, в методах молекулярной спектроскопии – с широкополосными.
Абсорбционные методы основаны на измерении поглощения веществом светового излучения. Различают молекулярно-абсорбционные и атомноабсорбционные методы анализа, которые отличаются способами возбуждения вещества.
Методы, основанные на измерении интенсивности света, излучаемого веществом, называют эмиссионными. К молекулярно-эмиссионным относят флуориметрию, к атомно-эмиссионным – эмиссионный спектральный анализ, в том числе пламенную фотометрию. Деление анализа на эмиссионный и флуоресцентный объясняется различным способом возбуждения вещества. В эмиссионном анализе частицы вещества возбуждаются в источнике света (пламени, газовом разряде и т. д.), куда вводится анализируемая проба. Во флуоресцентном анализе частицы вещества возбуждаются квантами электромагнитного излучения, которое направляют на анализируемую пробу от внешнего источника.
В молекулярном анализе используют также методы, основанные на измерении эффектов поляризации (рефрактометрия, интерферометрия и поля-
Химия. Учеб. пособие |
-194- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
риметрия), и методы, основанные на измерении интенсивности света, рассеянного или пропущенного суспензией вещества (фотонефелометрия, фототурбидиметрия).
По способу регистрации спектров различают оптические методы ана-
лиза визуальные, когда спектры наблюдают в видимой области спектра визуально или с помощью спектроскопов; фотографические, когда используют фотографическую пластинку или пленку для регистрации спектра; фотоэлектрические, когда используют фотоэлементы, фотоумножители и фотосопротивления.
Взаимодействие электромагнитного излучения с веществом можно рассматривать и как процесс возникновения аналитических сигналов, которые содержат информацию о свойствах исследуемого вещества. Частота сигнала отражает специфические свойства вещества, связанные с его строением, что дает возможность выполнять качественный анализ. Интенсивность сигнала связана с количеством анализируемого вещества.
Выполняя качественный атомный анализ, определяют элементы, входящие в состав анализируемой пробы. Молекулярный анализ позволяет установить природу индивидуального соединения, расшифровать структуру его молекул, определить, какие вещества входят в состав исследуемой смеси.
Концентрацию компонентов в исследуемом образце устанавливают количественным анализом. По способу выполнения количественные методы подразделяются на прямые и косвенные.
Впрямых методах снимают зависимость аналитического сигнала (показателя преломления, интенсивности поглощения, пропускания, излучения и т. д.) от концентрации. В косвенных оптических методах используют титрование и измеряемый аналитический сигнал применяют для определения объема в конечной точке титрования. Прямые методы анализа подразделяют на метод градуировочного графика, сравнения (стандартов, эталонов), добавок, расчетный метод, метод ограничивающих растворов.
Вкосвенных оптических методах анализа в процессе титрования происходит изменение концентраций реагирующих веществ, что сопровождается изменением оптического свойства, связанного с составом раствора. На основании полученных данных строят кривую титрования, форма которой зависит от типа протекающей реакции, природы реагирующих веществ, закономерности изменения измеряемой величины.
Ввидимой области спектра форма кривой титрования зависит от того, когда появилась окраска раствора (до точки эквивалентности или после неё)
икак изменялась её интенсивность в процессе титрования. Кривые титрования имеют вид нескольких прямых с точкой перегиба в конце титрования каждого компонента. Объем в конечной точке титрования находят на пересечении касательных, проведенных к различным участкам кривой титрования.
Происхождение спектров. Современное учение о спектрах электромагнитного излучения базируется на квантовой теории, согласно которой атомная или молекулярная системы являются устойчивыми лишь в определенных стационарных состояниях, соответствующих некоторой дискретной или непрерывной последовательности значений энергии Е. Изменение этой
Химия. Учеб. пособие |
-195- |
13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
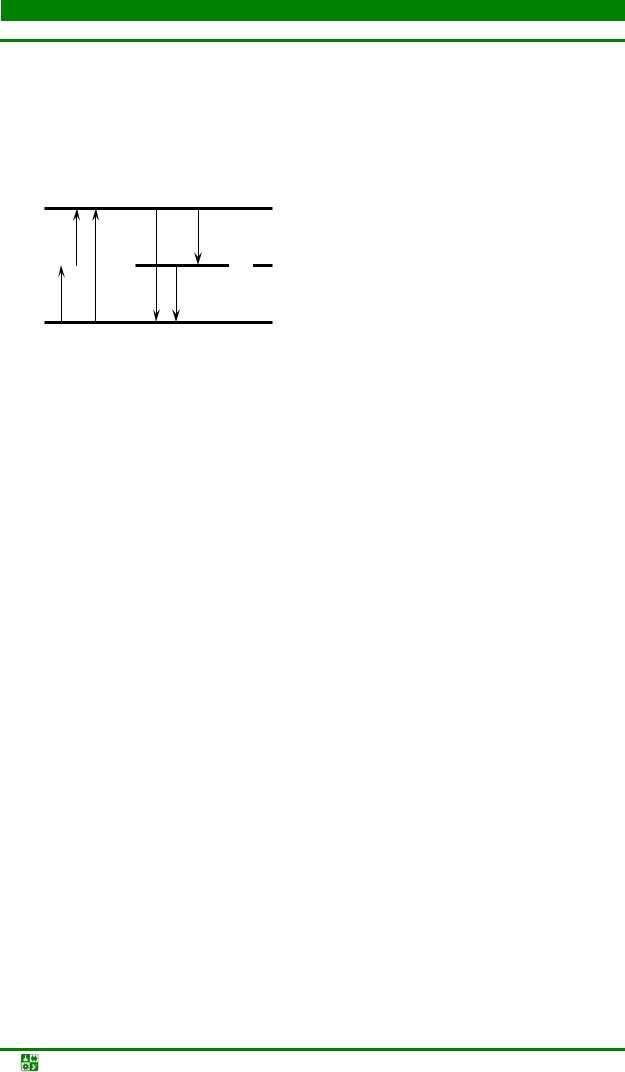
энергии связано со скачкообразным переходом системы из одного стационарного состояния в другое. При этом происходит либо поглощение, либо испускание кванта электромагнитного излучения. Условно энергетические состояния атомов и молекул обозначают в виде горизонтальных прямых – энергетических уровней (рис. 13.1).
E2 |
|
|
|
|
Поглощение |
||
|
|
|
|
E1 |
|
|
|
|
|
|
|
|
|
|
|
E0 |
|
|
|
|
|
а
Испускание
E2 |
|
|
|
|
|
γ2 |
|
|
|
υ′2 |
|
|
||
|
|
|
γ1 |
|
|
|
υ′1 |
|
|
||
|
||
|
|
|
E1 |
|
υ′ |
|
||
|
||
|
||
γ2 |
|
|
|
|
|
|
υ2 |
|
|
||
|
|
|
γ1 |
|
|
|
||
|
υ1 |
|
|
||
E0 |
|
|
|
||
|
υ |
|
|
||
|
||
|
б
Рис. 13.1. Схема энергетических уровней и электронных переходов в атомах (а)
имолекулах (б): Е – электронные уровни; υ – колебательные уровни;
γ– вращательные уровни
Частицы (атомы и молекулы), обладающие минимальным запасом внутренней энергии Е0, называют невозбужденными, а состояние, в котором они находятся – нормальным или основным. Путем внешнего воздействия частицам можно сообщить дополнительную энергию, поглотив которую, они переходят в возбужденное состояние с большим запасом энергии Е1, Е2 … Процесс поглощения энергии частицами вещества обозначают стрелками вверх, а процесс испускания – вниз.
Частота испускаемого или поглощаемого излучения определяется разностью энергии между орбиталями. Вещество состоит из множества атомов, способных переходить на разные энергетические уровни, испуская или поглощая фотоны разных частот. Совокупность всех фотонов одной и той же частоты составляет спектральную линию. Совокупность всех спектральных линий называют абсорбционным (при поглощении) или эмиссионным (при испускании) спектром.
Внутренняя энергия атомов и молекул изменяеться скачкообразно (дискретно). Для атома каждого элемента, молекул каждого вещества существует свой прерывный ряд энергетических состояний, поэтому они могут испускать и поглощать энергию только строго определенными порциями и имеют свой характеристический спектр.
Энергетическое состояние атома определяется в основном энергией электронных уровней (рис. 13.1, а). Каждому возможному переходу электронов между уровнями энергий соответствует спектральная линия, характеризующаяся определенной частотой и длиной волны. Поскольку разность энергий электронных уровней достаточно велика, атомный спектр состоит из отдельных спектральных линий. Наиболее вероятны переходы с первого возбужденного уровня Е1 на основной Е0; соответствующие им спектральные
Химия. Учеб. пособие |
-196- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
линии называются резонансными. Эти линии обладают наибольшей интенсивностью, и их чаще используют в практике количественного анализа.
Энергетическое состояние молекулы сложнее, чем у атомов. Наряду с движением электронов происходит колебание самих атомов и вращение молекул как целого. Поэтому в любом стационарном состоянии энергия молекул складывается из электронной, колебательной и вращательной энергий. Так же как и атом, молекула может существовать только в определенных энергетических состояниях, называемых энергетическими уровнями. Каждому электронному состоянию отвечают колебательные уровни, а каждому колебательному – вращательные уровни (рис. 13.1, б).
Переход электрона с одной орбитали на другую обязательно сопровождается изменением колебательного и вращательного состояний. Число энергетических состояний молекул велико, и энергии отдельных переходов бывают настолько близки, что распределить их невозможно. Поэтому в молекулярных спектрах спектральные линии с близкими частотами смещаются в одну полосу поглощения. Совокупность полос поглощения и излучения данного вещества называют его спектром.
Таким образом, в отличие от атомных спектров, состоящих из отдельных линий, молекулярные спектры состоят из совокупности полос. Характер поглощения или излучения (спектра) зависит от природы вещества, на этом основан качественный анализ. Для количественных измерений используют зависимость интенсивности светопоглощения от концентрации.
Атомный спектральный анализ основан на получении и изучении спектров излучения (эмиссии) или поглощения (абсорбции). Абсорбционные спектры проще эмиссионных, т. к. в них наблюдаются в основном резонансные линии. Оптические характеристические спектры наблюдаются, когда исследуемое вещество атомизировано и имеет малую плотность. Атомизацию проводят в высокотемпературных условиях: пламени горелки, электрической дуги, высоковольтной искры, высокочастотной плазмы, лазера.
Эмиссионная фотометрия пламени основана на измерении интенсивности излучения атомов, возбуждаемых нагреванием вещества в пламени. Интенсивность излучения I пропорциональна концентрации С ионов в растворе: I = k C. Задача определения состоит в выделении характерного для данного элемента излучения и измерения его интенсивности. Осуществляется это с помощью специального прибора – пламенного фотометра, регистрирующего с помощью светофильтров излучения в некоторой области спектра. Этот метод применяется для определения щелочных (калия, натрия), щелочноземельных (магния, кальция, стронция, бария) металлов, реже марганца и меди.
Атомно-абсорбционная спектроскопия основана на поглощении излу-
чения свободными (невозбужденными) атомами. В этом методе применяют атомизаторы различных видов: пламя, электротермические атомизаторы, тлеющий разряд, лазер. В качестве источника света используют источники, излучающие узкие полосы спектра: лампы с полым катодом, лампы с газовым разрядом низкого давления, безэлектродные радиочастотные лампы. При прохождении через пламя горелки спектр лампы ослабляется на опреде-
Химия. Учеб. пособие |
-197- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
ленную величину, по которой судят о количестве устанавливаемого элемента. Недостатком метода является трудность осуществления многоэлементного анализа, т. к. для каждого элемента нужен свой источник света.
Молекулярный спектральный анализ. В основе метода лежит избира-
тельное поглощение или излучение света вещества: при некоторых длинах волн электромагнитного излучения светопоглощение происходит интенсивно, т. к. энергия квантов равна энергии возбуждения молекулы.
Молекулярно-абсорбционные методы анализа основаны на измерении поглощения молекулами вещества электромагнитного излучения оптического диапазона. В зависимости от способа измерений, ширины полосы измеряемого излучения различают следующие молекулярно-абсорбционные методы:
колориметрия – сравнение окраски анализируемого и стандартного растворов проводят визуальным способом, пропуская через исследуемый раствор полихроматический свет;
фотоколориметрический метод – основан на измерении поглощения анализируемым веществом не строго монохроматического видимого диапазона светопотока, интенсивность которого измеряют с помощью фотоэлемента;
спектрофотометрия – изучает поглощение анализируемым веществом света с определенной длиной волны, т. е. монохроматического излучения.
В зависимости от области оптического диапазона длин волн различают анализ в ультрафиолетовой (УФ), видимой и инфракрасной (ИК) областях спектра. Возникновение спектров поглощения в ультафиолетовой и видимой областях спектра объясняется способностью электронов поглощать кванты света и переходить на более высокие энергетические уровни, поэтому такие спектры называют электронными. В инфракрасной области кванты света поглощают отдельные функциональные группы, атомы которых при этом изменяют энергетические уровни своих колебательных и вращательных движений, поэтому ИК-спектры называют молекулярными.
Люминесцентный метод анализа. Люминесценция – способность к самостоятельному свечению, возникающему под действием ультрафиолетовых, рентгеновских или радиоактивных лучей (фотолюминесценция, рентге-
нолюминесценция, радиолюминесценция). Свечение может прекращаться сра-
зу при исчезновении возбуждения (флуоресценция) или продолжаться определенное время после прекращения возбуждающегося воздействия (фосфоресценция). В основном используют явление флуоресценции. Качественный флуоресцентный анализ заключается в изучении спектральных характеристик флуоресценции (спектров излучения), количественный анализ основан на прямой зависимости интенсивности люминесценции Iл от концентрации при С < 10–5 моль/л.
Для измерении флуоресценции используют спектрофлуориметры и флуориметры. Свет от источника возбуждения через светофильтры попадает на кювету с раствором, испускаемое излучение проходит через вторичный светофильтр, фотоэлемент, где преобразуется в фототок, величина которого тем больше, чем больше концентрация раствора.
Химия. Учеб. пособие |
-198- |
13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
Нефелометрия основана на измерении интенсивности света, рассеянного суспензией частиц, находящихся в кювете. При пропускании света через кювету часть его поглощается, часть отражается и значительная часть рассеивается во всех направлениях. Нефелометрическое определение состоит в сравнении светорассеяния анализируемой суспензии с аналогичным показателем стандартного раствора. Интенсивность рассеянного света связана с размерами и количеством частиц или количеством вещества, находящегося в частицах. С помощью нефелометрии измеряют концентрацию веществ, нерастворимых в воде, но образующих стойкие суспензии.
Турбидиметрия основана на измерении количества света, поглощаемого неокрашенными суспензиями. При турбидиметрических определениях поглощение света суспензией измеряют подобно тому, как это делают в случае колориметрирования окрашенных растворов.
Рефрактометрический метод анализа основан на измерении зави-
симости коэффициента преломления от качественного и количественного состава исследуемой системы. Преломлением или рефракцией называется изменение прямолинейного распространения света при переходе из одной прозрачной среды в другую.
Согласно закону преломления для сред, оптически однородных, отношение синусов угла падения и преломления – величина постоянная и называется показателем или коэффициентом преломления. Коэффициент преломления определяется природой вещества, его химическим строением и зависит от длины волны, температуры, плотности, концентрации.
Показатель преломления чаще измеряют при длине волны 589,3 нм (желтая линия D в спектре натрия) и обозначают n20D. Величину показателя преломления измеряют приборами рефрактометрами. Измерив показатель преломления и плотность чистого растворителя, раствора и растворенного вещества, рассчитывают состав раствора.
Поляриметрический метод анализа основан на измерении угла вра-
щения плоскости поляризации луча света, прошедшего через оптически активную среду. Некоторые вещества в растворе способны вращать плоскость поляризации поляризованного света и называются оптически активными. Величина угла вращения плоскости поляризации прямо пропорциональна концентрации оптически активного вещества в растворе. Эту зависимость используют для количественного анализа. Для измерения оптической активности используют приборы поляриметры.
13.3.2. Электрохимическиеметодыанализа
Электрохимические методы анализа (табл. 13.2) основаны на изучении процессов, притекающих на поверхности электрода или в приэлектродном пространстве. Аналитический сигнал – электрический параметр (потенциал, сила тока, сопротивление), который зависит от концентрации определяемого вещества. Различают прямые и косвенные методы. В прямых – используют зависимость силы тока от концентрации определяемого компонента; в кос-
Химия. Учеб. пособие |
-199- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
венных – силу тока (потенциала) измеряют для нахождения конечной точки титрования (точки эквивалентности) определяемого компонента титрантом.
Классификация электрохимических методов анализа |
Таблица 13.2 |
||
|
|||
|
|
|
|
Метод |
Измеряемый параметр |
Условия |
|
измерения |
|||
|
|
||
Потенциометрия |
Электродный потенциал Е, В |
I = 0 |
|
Кондуктометрия |
|
I 1 000 Гц |
|
|
Удельная электропроводность χ, См см–1 |
||
Высокочастотная |
I 5–50 МГц |
||
|
|||
кондуктометрия |
|
||
|
|
||
Электрогравиметрия |
Масса m, г |
I = const или |
|
Е = сonst |
|||
|
|
||
Кулонометрия |
Количество электричества Q, Кл |
I = const или |
|
Е = сonst |
|||
|
|
||
Вольтамперометрия |
Сила тока I, мкА |
I = f(Еналож) |
|
Потенциометрия основана на определении зависимости между равновесным электродным потенциалом Е и активностью а или концентрацией С компонентов. Зависимость равновесного потенциала электрода от активности ионов выражается уравнением Нернста:
Е = Еo + RT ln aox ,
nF ared
где Ео – стандартный электродный потенциал, В; R – универсальная газовая постоянная, равная 8,313 Дж/(моль К); Т – температура, К; F – число Фарадея, F = 96 500 Кл; n – число электронов, участвующих в электродной реакции; aox, ared – активности окисленной и восстановленной форм. На практике используют концентрации (С), а не активности (а), между которыми существует зависимость a = γ C, где γ – коэффициент активности.
О концентрации определяемых ионов в растворе судят по потенциалу индикаторного электрода (электрод, реагирующий на концентрацию определяемых ионов). Величину потенциала этого электрода устанавливают, сравнивая с потенциалом другого электрода – электрода сравнения (электрод с постоянным потенциалом, нечувствительный к ионам раствора).
Кондуктометрия основана на том, что при определенной температуре электрическая проводимость раствора пропорциональна концентрации электролита. На практике используют метод градуировочного графика. Для его построения измеряют электропроводность раствора при нескольких концентрациях электролита. Определяют электропроводность анализируемого раствора и по градуировочному графику находят концентрацию электролита.
Электрогравиметрия основана на использовании процесса электролиза, в результате которого определяемое вещество выделяется на одном из электродов в виде осадка, по массе которого рассчитывается исходная концентрация вещества. Электролиз при постоянной силе тока применим для
Химия. Учеб. пособие |
-200- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
определения ионов металлов с положительным значением стандартного электродного потенциала. Электролиз при постоянном потенциале катода высокоселективен для определения двух- и трехзарядных ионов металлов.
Кулонометрия – группа методов, основанных на измерении количества электричества, необходимого для электрохимического превращения определяемого вещества. Кулонометрический анализ базируется на явлении электролиза. Прямая кулонометрия основана на поддержании постоянства электродного потенциала рабочего электрода на протяжении всего электрохимического процесса. Прямая кулонометрия применяется для определения любых электрохимически активных веществ: катионов металлов, анионов, которые могут давать малорастворимые соединения (галогениды), органических соединений. Кулонометрическое титрование отличается от прямой кулонометрии тем, что титрант образуется в электрохимической ячейке из добавленных в неё веществ. Титрование проводят при постоянной силе тока. Для определения точки эквивалентности используют различные способы: визуальный, индикаторный, инструментальный (рН-метрия, амперометрия, спектрофотометрия).
Вольтамперометрия основана на изучении поляризационных или вольтамперометрических кривых (зависимостей силы тока от напряжения). Электролиз проводят с использованием легко поляризуемого электрода с небольшой поверхностью. Вольтамперометрический анализ используют для анализа неорганических и органических веществ, способных к электрохимическим превращениям.
Полярография. При анализе испытуемый раствор подвергают электролизу в ячейке прибора – полярографа. Полярограф автоматически записывает вольт-амперную кривую, показывающую изменение силы диффузного тока с повышением напряжения. По характеру кривой судят о присутствии тех или иных катионов в растворе и об их количественном содержании. Полярографию применяют также для определения конечных точек при титровании. С помощью полярографии в технических образцах определяют примеси металлов порядка 0,001 % с точностью до 1 %.
13.3.3. Хроматографическиеметодыанализа
Хроматографический метод – физико-химический метод разделения компонентов сложных смесей газов, паров, жидкостей или растворенных веществ, основанный на использовании сорбционных процессов в динамических условиях. В простейшем виде эти условия осуществляются при прохождении раствора, содержащего растворенные вещества, через колонку со слоем сорбента. Вследствие различной сорбируемости компонентов смеси происходит их разделение по длине колонки за счет многократного повторения сорбции, десорбции и других процессов.
Химия. Учеб. пособие |
-201- |
13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
Принцип |
Сорбционный |
|
|
|
|
Среда |
Газовая |
|
|
|
|
Жидкостная |
|
|
|
|
Газожидкостная |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Механизм |
Молекулярная |
|
Ионообменная |
|
|
Осадочная |
|
Распределительная |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Форма |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Колоночная |
|
|
|
Капиллярная |
|
|
|
Тонкослойная и бумажная |
|
||||||
применения |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
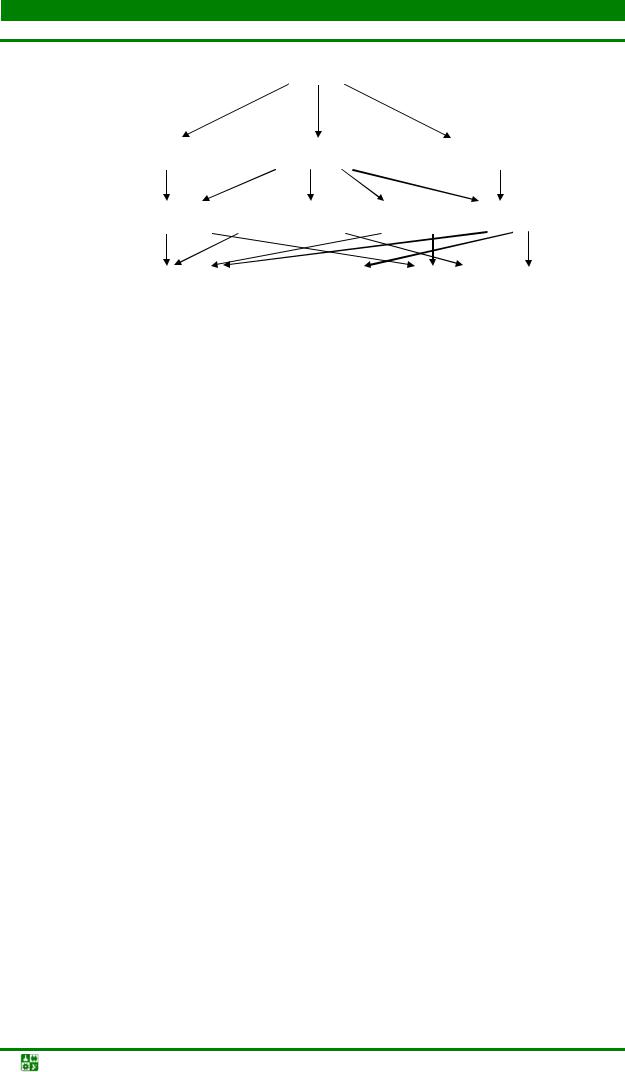
Рис. 13.2. Классификация хроматографических методов
В молекулярной хроматографии разделение осуществляется за счет молекулярных сил взаимодействия на границе твердой и жидкой фаз. Эти силы возникают между молекулами на поверхности сорбента и молекулами, растворенными в проходящей через колонку жидкости или газовой смеси. При этом на поверхности твердой фазы осуществляется сорбционный процесс при участии сил межмолекулярного притяжения, дипольного взаимодействия и поляризационных. При наличии в смеси веществ с различными сорбционными взаимодействиями процесс будет протекать с разделением смеси на хроматографический колонке. Такая хроматография называется адсорбционной.
К молекулярной относится и газовая хроматография. Она включает все методы, в которых одна фаза твердая, другая – газообразная. Обычно стационарная фаза – твердый сорбент, а подвижная – смесь газов. В случае газожидкостной хроматографии жидкая фаза находится на твердом носителе, а в качестве подвижной фазы выступает инертный газ с малой концентрацией исследуемого вещества. Образование хроматограмм происходит вследствие различия коэффициентов распределения веществ между неподвижной жидкой и подвижной газообразной фазами.
Важнейшие сорбенты адсорбционной хроматографии: карбонат кальция, оксид алюминия, силикагель, активированный уголь, инсулин, сахароза, крахмал, целлюлоза. В качестве растворителей используются пентан, гексан, петролейный эфир, бензин, бензол, хлороформ, четыреххлористый углерод, хлорбензол, фенолы и т. д.
Силы взаимодействия могут возникать и при контакте двух несмешивающихся жидких фаз. В практике обычно одна из жидких фаз закрепляется на поверхности твердого носителя, т. е. становится неподвижной. Вторая жидкая фаза подвижна. Распределение веществ происходит между этими фа-
зами. Сам метод носит название распределительной хроматографии. Ско-
Химия. Учеб. пособие |
-202- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
рость перемещения разделяемых веществ вдоль колонки определяется значениями их коэффициентов распределения.
Распределительная хроматография в колонке основана на распределе-
нии вещества между двумя жидкими фазами, одна из которых стационарная, а другая − подвижная. Анализируемая смесь вносится в виде раствора в подвижном растворителе в колонку. Затем колонка промывается чистым растворителем. При этом скорость движения компонентов по колонке тем больше, чем ниже коэффициент распределения KD:
KD = Cн ,
Cп
где Сн – молярная концентрация определяемого вещества в неподвижной фазе; Сп – то же в подвижной фазе. Коэффициент распределения зависит от природы вещества и растворителя и температуры.
Можно показать, что между коэффициентом распределения и величиной объемов подвижного Vп и неподвижного Vн растворителей, приходящихся на единицу объема колонки, существует зависимость:
KD = Vп ( 1 −1) ,
Vн R
где R – величина смещения зоны на единицу объема подвижного растворителя (при R = 1 вещество не задерживается в колонке, а движется с подвижным растворителем).
Величина смещения зоны на единицу объема подвижного растворителя определяется по формуле
R = |
A |
, |
Aп + KDAн |
где А – площадь поперечного сечения колонки,
А = Ai + Aп + Aн,
где Аi – площадь поперечного сечения носителя (инертного твердого вещества); Ап – то же подвижной фазы; Ан – площадь поперечного сечения неподвижной фазы.
В бумажной хроматографии капля исследуемого вещества наносится на бумагу и этот конец бумаги помещается в растворитель, который под действием капиллярных сил поднимается вверх. Когда растворитель подойдет к месту нанесения смеси веществ, происходит распределение отдельных компонентов между подвижной и неподвижной фазами, основанное на различии их коэффициентов распределения. При этом за счет многократного повторе-
Химия. Учеб. пособие |
-203- |
13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
ния акта распределения между двумя фазами образуются отдельные зоны (пятна) индивидуальных веществ. Перемещение полосы растворенного вещества описывается величиной Rf :
|
Скорость движения зоны |
|||||||
Rf = |
|
|
|
. |
||||
Скорость движенияфронта растворителя |
||||||||
Это уравнение преобразуется к виду |
|
|
|
|||||
|
Rf = |
RAп |
= |
|
|
Aп |
, |
|
тогда |
A |
Aп + KD Aн |
|
|||||
|
|
|
|
|||||
|
|
Aп |
|
|
|
|
|
|
|
|
|
1 |
|
|
|
||
|
KD = |
Ан |
|
|
−1 . |
|
|
|
|
|
|
|
|||||
|
|
|
Rf |
|
|
|
||
Это уравнение позволяет определить KD при известных Rf и соотношении Ап/Ан, которое принимают равным отношению объемов растворителя и водной фазы в полоске бумаги. Экспериментально его можно найти, зная вес бумажной полосы до хроматографирования с учетом содержания воды в ней и вес после хроматографирования. Rf можно определить экспериментально
(рис. 13.3):
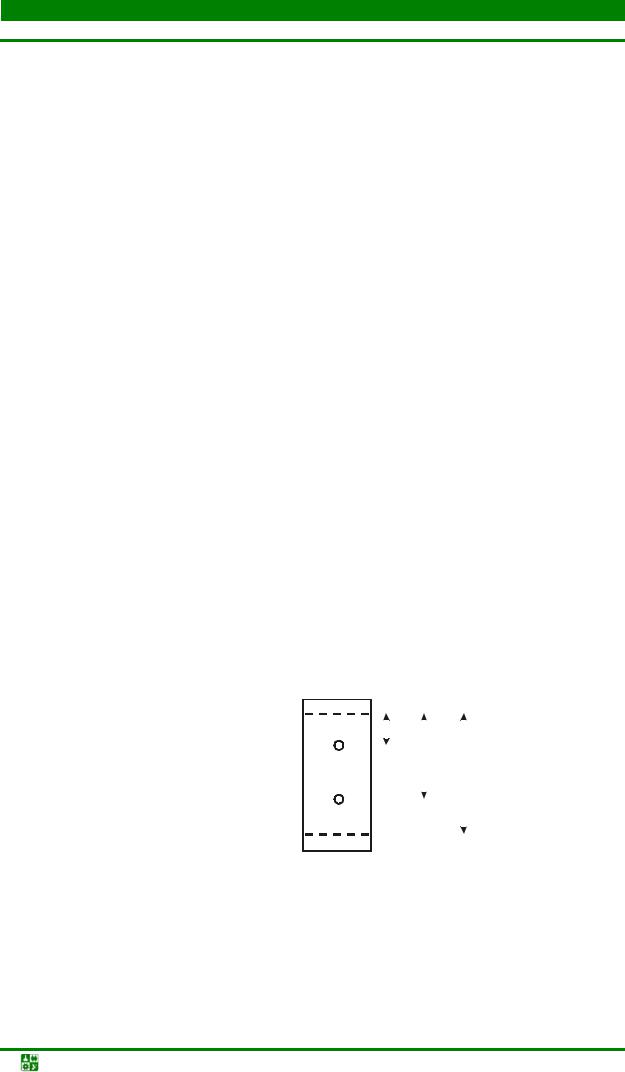
Rf = l/L,
где l – расстояние, пройденное веществом; L – расстояние, пройденное растворителем.
Разделение двух веществ с Rf и Rf" практически возможно, если Rf' > Rf" и Rf' − Rf" > 0,1.
начальная |
ия |
|
|
|
|
|
|
|
|
|
|||
Начальная линия |
|
|
|
ll1 |
|
|
|
|
|||||
|
' |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
1 |
|
|
|
|
||
fR |
1= l |
|
/L |
1 |
|
|
|
|
l |
|
|
|
|
R ' = l /L |
1 |
|
|
|
|
|
l22 |
|
|
||||
|
f |
|
|
|
|
|
|
|
L |
||||
|
'' |
|
|
/L |
2 |
|
|
|
|
|
|
L |
|
RfR f 2= l2 |
|
|
|
|
|
|
|
|
|||||
'' = l |
/L |
|
|
|
|
|
|
|
|
|
|
|
|
Фронтфронтрастворителярас теля |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|||||
Рис. 13.3. Схема разделения двух ионов на бумаге
иэкспериментальное определение Rf
Влюбом методе бумажной хроматографии разделение проводят в герметически закрытых камерах, атмосфера которых насыщена парами исполь-
Химия. Учеб. пособие |
-204- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
зуемого растворителя. Существуют разные методы получения бумажных хроматограмм.
Качественный анализ неорганических веществ проводят по характерной окраске зон. В случае если зоны невидимы, их проявляют специфическими реагентами.
Другой способ основан на измерении Rf для каждого исследуемого вещества в определенном растворителе.
В бумажной хроматографии возможно количественное определение веществ. В качестве носителя используется специальная хроматографическая бумага. Основные растворители – спирты, простые эфиры, кетоны, эфиры органических кислот, органические основания, трибутилфосфат, хлороформ. Как правило, их насыщают водой, минеральными или органическими кислотами.
Тонкослойная хроматография. Для хроматографии в тонком слое используются различные сорбенты: оксид алюминия, силикагель, ионообменные смолы. Перемещение жидкости в слоях происходит так же, как в колоночной хроматографии. Это позволяет использовать принципы адсорбционной, распределительной и ионообменной хроматографии. В методе используется величина Rf, которая определяется так же, как в бумажной хроматографии. Она зависит от активности и качества сорбента, толщины слоя носителя, качества и природы растворителя, количества нанесенного вещества и способа работы. Для идентификации веществ определяют Rf для анализируемого вещества и свидетеля, которые наносят на эту же пластинку вместе с исследуемой смесью веществ. Положение пятен на хроматограмме выражают в виде отношения
RS = Rfu ,
Rfe
где Rfu – величина, характеризующая положение пятна исследуемого вещест-
ва; Rfe – то же пятна-свидетеля.
Ионообменная хроматография – разделение веществ, основанное на обратимом обмене ионов, содержащихся в растворе, на ионы, входящие в состав ионообменника. Образование хроматограмм при этом происходит за счет различной способности к обмену ионов хроматографируемого раствора.
Осадочная хроматография – распределение веществ вследствие образования малорастворимых осадков в определенном порядке, который обуславливается их растворимостью. По мере фильтрации раствора через оса- дочно-хроматографическую колонку, содержащую осадитель, многократно повторяются элементарные процессы образования и растворения осадков, что обеспечивает разделение веществ. Растворимость осадков и произведе-
Химия. Учеб. пособие |
-205- |

13.ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
13.3.Физико-химические методы анализа
ние растворимости, как характеристика этого свойства осадков, выступает как основной закон осадочной хроматографии. Возможность повторения элементарного процесса обеспечивается закреплением осадков в месте их образования, в противном случае осадки будут сползать вниз и хроматограмма не образуется.
Контрольныевопросыизадания
1.Как классифицировать методы анализа в аналитической химии? Что является основой каждого метода?
2.Какие аналитические реакции используются в качественном химическом анализе для обнаружения ионов?
3.Чем отличаются специфические и избирательные реакции?
4.Каково соотношение между открываемым минимумом mmin и предельным разбавлением сmin?
5.Что является аналитическим сигналом? Какова зависимость между интенсивностью аналитического сигнала и содержанием анализируемого компонента?
6.Каковы цели и задачи качественного анализа ?
7.Каким образом устраняются мешающие ионы в качественном ализе?
8.В какой среде восстанавливаются только ионы Hg2+ без мешающих ионов Bi3+ и Ag+? Привести уравнения реакций.
9.На чём основан принцип классификации катионов по кислотнощелочному и сульфидному методам на аналитические группы?
10.Какая реакция используется одновременно для открытия ионов Ва2+
иSO 24− ? Записать её в молекулярной и ионно-молекулярной форме. Указать
цвет осадка.
11.Какая реакция является одновременно характерной для открытия ионов Ag+ и Cl–? Записать её в молекулярной и ионно-молекулярной форме. Указать цвет осадка.
12.Какой объем 0,1 М раствора Н2SО4 необходим для осаждения Ва2+ из навески препарата массой 2,35 г, содержащего 92 % ВаСl2?
13.Привести уравнения образования нерастворимых гидроксидов, карбонатов, сульфидов, сульфатов, фосфатов различных катионов, используемых в качественном анализе, расставить коэффициенты:
а) H2O2 + Cr3+ + OH– = |
б) Bi3+ + Sn2+ + OH– = |
14. Что является основой классификации анионов ? Написать реакции открытия катионов К+ и NH+4 в молекулярной и ионно-молекулярной форме.
Химия. Учеб. пособие |
-206- |

13. ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
Контрольные вопросы и задания
15.Какая реакция является одновременно характерной для открытия ионов Pb2+ и J–? Записать её в молекулярной и ионно-молекулярной форме. Указать цвет осадка.
16.Как обнаружить присутствие в растворе Mg2+ и Al3+. Записать реакции в молекулярной и ионно-молекулярной форме.
17.Какие реакции являются характерными для обнаружения ионов Te2+
иFe3+? Записать их в молекулярной и ионно-молекулярной форме, указать изменение окраски растворов.
18.Записать реакции открытия анионов Cu2+, Co2+, Ni2+ гидроксидом аммония в молекулярной и ионно-молекулярной форме, указать в каждом случае изменение окраски раствора.
19.Записать реакции открытия анионов РО34− , SiO 32− в молекулярной и ионно-молекулярной форме. Указать характерные особенности этих реакции.
20.Записать реакции открытия ионов NO 3− , NO −2 , Cu3СОО– в молеку-
лярной и ионно-молекулярной форме. Указать характерные особенности этих реакции.
21.На каких явлениях основаны оптические методы анализа?
22.Раскрыть сущность метода эмиссионной фотометрии пламени.
23.Наизмерениикакойвеличиныоснованатомно-адсорбционныйанализ?
24.Какие величины используются для качественного и количественного анализа в спектрофотометрии?
25.На измерении какой величины основан люминесцентный метод
анализа?
26.На чем основан качественный анализ в флуориметрии?
27.Почему происходит преломление светового луча на границе разде-
ла сред?
28.Какие вещества называются оптически активными и на чем основан их анализ?
29.На чем основаны потенциометрические методы анализа (прямые и косвенные)?
30.На измерении какой величины основан кондуктометрический метод
анализа?
31.В чем сущность высокочастотного титрования?
32.На использовании какого электрохимического явления основан метод вольтамперометрии?
33.Чем определяется вид кривой амперометрического титрования?
34.Какие методы анализа относятся к хроматографическим и как они классифицируются?
Химия. Учеб. пособие |
-207- |

13. ХИМИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
Контрольные вопросы и задания
35. Как проводят качественный и количественный анализ в газовой, жидкостной, бумажной и тонкослойной хроматографиях?
Компетенциистудента
Изучив содержание этой темы, студент должен:
знать схемы аналитического определения; способы подготовки пробы; количественные измерения, аналитический сигнал; требования, предъявляемые к анализу; классификацию методов анализа; специфические, избирательные реакции; чувствительность характерных реакций; макро-, по- лумикро-, микрометоды анализа; технику разделения и идентификации катионов; способы устранения мешающих катионов; этапы гравиметрического анализа; требования к гравиметрической форме; расчет гравиметрического фактора; титриметрический анализ; стандартизацию растворов титрантов; происхождение спектров; оптические методы анализа (эмиссионную фотометрию пламени, атомный и молекулярный спектральные анализы, люминесцентный, рефрактометрический и поляриметрический анализы); электрохимические методы анализа (потенциометрию, кондуктометрию, электрогравиметрию, кулонометрию, вольтамперометрию); хроматографические методы анализа (молекулярную, ионообменную, осадочную и распределительную хроматографию);
уметь отбирать и усреднять пробы; растворять, вскрывать пробы; рассчитывать интенсивность аналитического сигнала; определять класс методов анализа; вычислять открываемый минимум mmin; осаждать ионы, используя различные реагенты; изменять кислотность среды; окислять, восстанавливать мешающие ионы; применять этапы гравиметрического определения; вычислять гравиметрический фактор, фактор эквивалентности, молекулярную массу эквивалентов; рассчитывать результаты различного титрования, результаты по известному титру.
Химия. Учеб. пособие |
-208- |