

27. См документ 0048.
28.Три физических (релаксационных) состояния аморфных полимеров. Температура стеклования. Для аморфных полимеров в зависимости от температуры (и величины механического напряжения) возможны три физических (деформационных) состояния: стеклообразное, высокоэластическое и вязкотекучее. Практическое применение полимеров определяется тем, в каком из этих состояний находится данный полимер при температуре его использования.
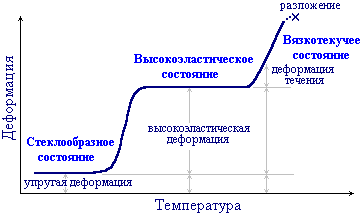
Для стеклообразных полимеров характерны относительно небольшие упругие (обратимые) деформации (1-10%). Причем полимерные стекла отличаются повышенной прочностью от низкомолекулярных стеклообразных тел, которые разрушаются при деформировании уже на 0,1-1%. Полимеры в стеклообразном состоянии применяются в производстве пластмасс.
Высокоэластические полимеры способны обратимо деформироваться на сотни процентов. В высокоэластическом состоянии в условиях эксплуатации находятся все каучуки. Это состояние характерно лишь для полимеров.
В вязкотекучем состоянии полимер ведет себя как очень вязкая жидкость, которая под действием силы проявляет необратимую деформацию (деформацию течения). Это состояние реализуется обычно при повышенных температурах и используется для переработки полимеров в изделия.
Аморфное состояние. Нахождение аморфных полимеров в одном из трех физических состояний (стеклообразное, высокоэластическое или вязкотекучее) зависит от температуры. При нагревании или охлаждении аморфные полимеры могут обратимо переходить из одного физического состояния в другое, причем такие переходы не являются фазовыми, так как во всех трех состояниях сохраняется аморфная структура. Границами раздела физических состояний служат две температурные точки:
- температура стеклования Тс, являющаяся средним значением температурной области перехода из стеклообразного в высокоэластическое состояние;
- температура текучести Тт как среднее значение области перехода из высокоэластического в вязкотекучее состояние.
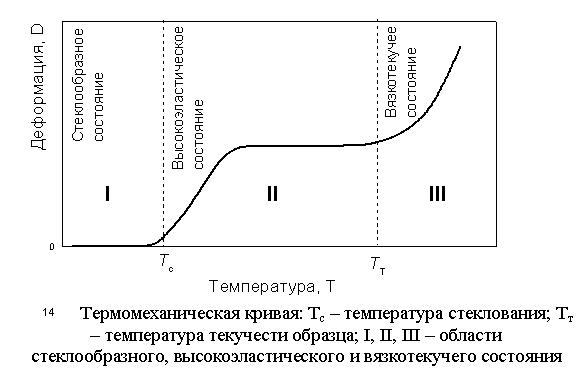
Для определения температур стеклования и текучести используют дилатометрический или термомеханический методы. Чаще используют метод термомеханических кривых, который состоит из двух этапов.
Первый этап – проведение эксперимента и получение данных. К образцу подвешивают груз. Затем образец с грузом помещают в печку. Температура в печке медленно повышается. Измеряются температура печки Т и удлинение образц D:
D=Di - Do
где Do - начальная длина образца; Di длина образца в момент i-го замера.
Второй этап – графическое представление полученных результатов – построение термомеханической кривой (рис. 14).
 В
области I удлинения
образца нет; в области II происходит
удлинение образца; в области III образец
удлиняется, пока не рвется.
В
области I удлинения
образца нет; в области II происходит
удлинение образца; в области III образец
удлиняется, пока не рвется.
Типичная термомеханическая кривая имеет два перегиба, являющихся температурными областями переходов. Тс и Тт находят как проекции точек пересечения прямолинейных участков кривой.
Кристаллическое состояние характеризуется температурами кристаллизации (Ткр) и плавления (Тпл).Температура кристаллизации Ткр – температура образования устойчивых кристаллических зародышей (находится в области между температурой стеклования и температурой текучести: Тс < Ткр < Тт). Температура плавления Тпл – температура плавления кристаллической фазы. Она может быть ниже или выше температуры текучести Тт. Если ниже – кристаллический полимер при плавлении переходит в высокоэластическое состояние; если выше – в вязкотекучее. Температура кристаллизации всегда ниже температуры плавления: Ткр< Тпл .
На температуры переходов значительное влияние оказывают молекулярная масса и химическое строение полимера. Так, жесткоцепные полимеры не имеют высокоэластического состояния. У них температура текучести совпадает с температурой стеклования, и ее в этом случае называют температурой размягчения. Некоторые полимеры, например целлюлоза, политетрафторэтилен и др., не переходят в вязкотекучее состояние, так как их гипотетическая температура текучести выше температуры разложения.
При низких температурах и стеклообразные, и кристаллические полимеры способны разрушаться при малых деформациях. Такой вид разрушения называется хрупким, а температура, соответствующая ему –температурой хрупкости Тхр. Температура хрупкости находится ниже температуры стеклования: Тхр< Тс. Ниже температуры хрупкости полимеры эксплуатировать нельзя.
Каждое физическое состояние характеризуется своим типом деформации (табл. 3).
Высокоэластическая деформация имеет релаксационную природу. Любой релаксационный процесс характеризуется временем релаксации, необходимым для перестройки структурных элементов. В реальных полимерах существует спектр времен релаксации, соответствующий перегруппировкам звеньев и участков цепей. Из-за релаксационного характера высокоэластической деформации времена достижения равновесного состояния могут быть очень значительны.
29. Свойства аморфных полимеров в стеклообразном состоянии. Теории стеклования
ТЕКЛООБРАЗНОЕ СОСТОЯНИЕ ПОЛИМЕРОВ, состояние, в котором полимер обладает физ.-мех. свойствамитвердого тела (не способен к текучести и высокоэластич. деформации. имеет малые значения коэф. термич. расширения и сжимаемости) при сохранении структурной неупорядоченности, присущей жидкому или высокоэластич. состоянию. Полимерные стекла, состоящие из длинных макромолекул с заторможенным внутр. вращением, при "расстекловыва-нии" переходят не в текучее (как низкомол. вещества), а в высокоэластич. состояние.
Стеклование обычно трактуется как кинетич. явление, когда при понижении температуры или повышении частоты воздействия характерные времена перестройки молекулы. ответственной за проявление высокоэластичности, оказываются сравнимыми с временем опыта или периодом воздействия. Физ. свойства полимера в стеклообразном состоянии определяются соотношением энергий межмол. взаимодействий и энергии теплового движения, а также плотностью упаковки (т.е. величиной своб. объема) полимера, конформац. свойствами полимерных цепей. Существует трактовка стеклования как квазиравновесного термодинамич. перехода 2-го рода, при котором в области низких температур обращается в нуль или резко падает энтропия переохлажденной полимерной жидкости (из-за уменьшения числа конформац. состояний макромолекул). Флуктуац. теории стеклования учитывают локальную структурную и динамич. гетерогенность полимера. Согласно кластерным концепциям, полимер в стеклообразном состояниисодержит отдельные изолир. подвижные участки (дефекты), обусловленные флуктуациями плотности или конформац. структуры цепей. При температурах выше стеклования температуры Тс эти участки сливаются в макроскопич. кластеры, что приводит к росту макроско-пич. мол. подвижности и переходу в высокоэластич. состояние. В застеклованном полимере сохраняются, как правило, различные формы локальной подвижности, связанные с движением боковых групп или малых фрагментов основных цепей.
Вязкость полимеров в стеклообразном состоянии составляет 1013-1014 Па·с, а характерные времена сегментального движения вблизи Tс превышают 102-103 с. При заданном давлении значения Tс полимеров намного выше Тссоответствующих низкомол. мономерных жидкостей. В случае гиб-коцепных полимеров Тс растут с увеличением мол. массы, стремясь к некоторому предельному значению, когда контурная длина цепей превышает длину кинетич. сегмента, а вклад концевых групп в величину своб. объема и мол. подвижность становится пренебрежимо малым. Значение Тс полимеров сильно зависит от давления, скорости нагрева (или охлаждения), частоты периодич. воздействия; наблюдаются гистерезисные явления. При образовании полимерных сеток (благодаря поперечным хим. связям) Тс возрастают. Наличие водородных и др. сильных межмолекулярных (полярных, ионных) связей также ведет к повышению Тс. При добавлении низкомол. примесей (пластификации) Тс полимера падает. Величина Тс сополимеров (статистических и блоксополимеров) зависит от состава, совместимости и строения сомономеров.
30.Аморфные полимерные стёкла. Упругая и вынужденно-эластическая деформация полимерных стёкол. Для стеклообразного состояния характерна упругая деформация:
![]()
где ε – модуль упругости (модуль Юнга); σ – механическое напряжение (измеряется на динамометре); D – относительная деформация (удлинение).
При больших напряжениях в определенном температурном интервале стеклообразные полимеры способны подвергаться значительным деформациям – до нескольких сотен процентов. Такие деформации близки по своей природе к высокоэластическим, поэтому они были названы высокоэластическими (А.П. Александров,1944 г.), а само явление – явлением вынужденной эластичности.
Зависимость напряжения σ от деформации D показана на рис. 19.
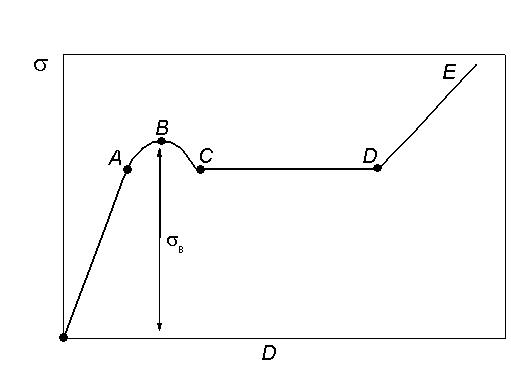
Кривую можно разделить на несколько участков.
На участке oA соблюдается линейная зависимость напряжения от деформации, что соответствует обратимой упругой деформации.
На участке АВ тангенс угла наклона кривой к оси абсцисс с увеличением напряжения уменьшается. Это связано с началом развития в образце вынужденной эластичности. Под действием значительных внешних напряжений макромолекулы разворачиваются и ориентируются в направлении действия силы. В точке B(максимум на кривой) напряжение достигает значения, соответствующего пределу вынужденной эластичностиσв.
На участке BC наблюдается спад напряжения. Это происходит из-за начала образования шейки. В точке Спроцесс формирования шейки заканчивается.
На отрезке СD происходит удлинение шейки за счет соседних, мало деформированных участков образца. На этом этапе σ = соnst, поэтому прямая СD параллельна оси абсцисс. В точке D весь образец переходит в шейку, т.е. его толщина становится равной толщине шейки. Рост шейки прекращается.
В области DE происходит дальнейшее удлинение образца как единого целого. Здесь, как и на первой стадии оА, зависимость напряжения от деформации линейна и подчиняется закону Гука.
У некоторых полимеров (например, нитрат целлюлозы) высокоэластическая деформация не сопровождается образованием шейки, в этом случае на деформационной кривой отсутствует максимум.
Вынужденная эластичность, как и высокоэластичность, носит релаксационный характер, т.е. зависит от времени и скорости деформации. Чем меньше время воздействия и больше скорость деформации, тем больше напряжение, а значит, выше значение предела вынужденной эластичности σв. При этом область вынужденной эластичности сокращается вплоть до исчезновения, когда образец разрушается ниже σв.
Аналогичная зависимость наблюдается с понижением температуры (рис. 20), что отвечает принципу температурно-временной суперпозиции: увеличение частоты действия силы эквивалентно понижению температуры (рис. 21). С повышением температуры уменьшается значение σв , что видно из рис. 20. При Т =Тс предел вынужденной эластичности становится равным нулю: σв = 0. При понижении температуры до значения, когда она становится равной температуре хрупкости Т = Тхр, напряжение, необходимое для перегруппировки сегментов под действием
силы, достигает значения разрушающего напряжения σхр (σв = σхр). При этом происходит хрупкое разрушение материала.
Таким образом, температуру хрупкости можно определить как температуру, ниже которой полимер не проявляет вынужденной эластичности.
31.Пластификация полимеров. Механизмы пластификации. Правила объемных и мольных долей. Смещение температуры стеклования полимеров в область более низких температур под действием введенных веществ называетсяпластификацией. Пластификаторы – вещества, способные смещать температуру стеклования полимеров в область более низких температур. Однако пластификаторы вводят не только для снижения температуры стеклования, но и для снижения модуля упругости, температуры текучести, вязкости полимеров, повышения эластичности, деформируемости материала при воздействии механических усилий во всех трех физических состояниях – стеклообразном, высокоэластическом и вязкотекучем. Но критерием, определяющим наличие явления пластификации и его эффективности, является смещение температуры стеклования полимеров в область более низких температур.