

10.1. Оптичні методів в аналізі об’єктів довкілля
Оптичні методи аналізу ґрунтуються на залежності характеристик електромагнітного випромінювання, таких як інтенсивність забарвлення розчину, його оптична густина та інші, від вмісту речовин, які цими методами визначають. Основою фізико-хімічної суті оптичних методів є взаємодія електромагнітного випромінювання з атомами чи молекулами речовини, яка супроводжується випромінюванням, поглинанням, розсіюванням, одночасним поглинанням і розсіюванням. Для збудження електронних рівнів необхідна енергія ультрафіолетової (200-400 нм) та видимої (400-700 нм) ділянок спектра електромагнітного випромінювання; для збудження коливальних рівнів – енергія інфрачервоної ділянки (700 – 75000 нм), а для збудження обертальних рівнів – енергія далекої інфрачервоної та мікрохвильової ділянок (75000-100000 нм). Якщо збуджена молекула може виділити випромінювання, енергія якого менша за поглинуту, такий вид розрядження збуджених молекул лежить в основі люмінесцентного аналізу.
10.1.1. Застосування спектрофотометричного аналізу
У спектрофотометричних методах аналізу поглинання світла ультрафіолетової чи видимої ділянки спектра збуджує електронну систему молекул. Збуджений стан нестійкий і за короткий час (~10-8 с) надлишок енергії виділяється в тій чи іншій формі. Поглинута енергія може призвести до зміни хімічного стану речовини. Так KMnO4 під дією світла розкладається, комплекс [Fe(SCN)6]3- є нестійкий. Переважна частина енергії перерозподіляється по окремих енергетичних рівнях молекули та перетворюється в тепловий рух, при цьому зростає внутрішня енергія.
Теоретичні основи спектрофотометричних методів аналізу описані у відповідній літературі з аналітичної хімії. Здатність поглинати світло залежить від природи речовини та концентрації. В основі спектрофотометричного методу аналізу є основний закон світлопоглинання (Бугера-Ламберта-Бера):
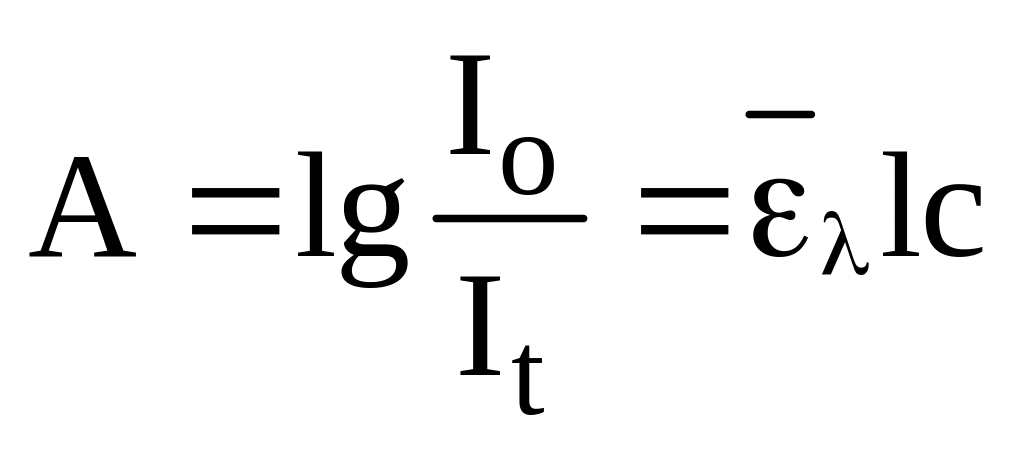
де А – оптична густина розчину; С – концентрація розчину, (моль/л);
Іо.
І – інтенсивність світлового потоку
падаючого та того, який пройшов через
розчин; l – товщина шару розчину, см;
![]() –
молярний коефіцієнт поглинання,
л×моль-1×см-1.
–
молярний коефіцієнт поглинання,
л×моль-1×см-1.
Чим більший молярний коефіцієнт поглинання, тим меншу концентрацію речовини можна визначити. Найменша визначувана концентрація становить Сmin=1106 моль/л.
Чим вужча смуга поглинання речовини в електронному спектрі, тим більш селективне визначення, а чим більша інтенсивність поглинання, тим чутливіший метод.
Точність фотометричних методів залежить від особливостей реакцій, приладів та ін., відносна похибка становить приблизно 1 2%.
Відносну похибку визначення концентрації обчислюють як:
![]() .
.
Визначено, що її величина найменша при світлопропусканні Т=0,37, тобто, оптимальне значення оптичної густини становить А=0,435.
Спектрофотометричні методи аналізу найчастіше застосовують для визначення неорганічних речовин у довкіллі. Серед органічних речовин цим методом визначають формальдегід, кетони, органічні аміни, сульфуровмісні сполуки, пестициди. Нажаль, іноді чутливість та селективність цих методів недостатня. Тому часто перед визначенням інгредієнта проводять концентрування проби та відділення визначуваного елемента від іонів, які заважають.
Деякі визначення спектрофотометричними методами, були приведені вище у розділі 9, а деякі описані в розділі 13.2, присвяченому дослідженню органічних речовин. Тут наведено коротку характеристику найчастіше використовуваних фотометричних визначень об’єктів довкілля без деталізації методики.
РО43- з молібденовою рідиною у кислому середовищі утворює жовту гетерополікислоту
H3PO4 +12 H2MoO4 = H3[PMo12O40] + 12H2O,
з
якої відновленням молібдену одержують
продукт синього кольору (![]() =18,5103).
Як
відновники використовують, аскорбінову
кислоту, Sn2+,
SO32-.
Колір розчину розвивається протягом
декількох хвилин і стійкий тривалий
час. Стійкість комплексу та інтенсивність
забарвлення залежать від рН.
=18,5103).
Як
відновники використовують, аскорбінову
кислоту, Sn2+,
SO32-.
Колір розчину розвивається протягом
декількох хвилин і стійкий тривалий
час. Стійкість комплексу та інтенсивність
забарвлення залежать від рН.
AsO43-
та SiO32-
визначають у формі гетерополікислот.
Розчинна моносилікатна кислота при
рН=1,6 реагує з молібдатною кислотою з
утворенням розчину β-силіціймолібдатної
гетерополікислоти H4(SiMo12O40),
яка аналогічно відновлюється до сполуки
синього кольору,
![]() =20103.
Для
H3[AsMo12O40]
=20103.
Для
H3[AsMo12O40]
![]() =25.Заважають
визначенню фосфати, германати.
=25.Заважають
визначенню фосфати, германати.
Аскорбінову кислоту визначають як відновник фосфоромолібдату (NH4)3[PMo12O40] – утворюється продукт синього кольору.
Фотометричне визначення сполук нітрогену (NH4+, сполук з пептидними зв’язками, NO2-, NO3-), фториду, хлориду, сульфіду описане в розділах 9.3, 9.4.
SO42- можна визначити йодид-йодатним методом, при якому сульфат витісняє йодат із барій йодату, а йодат з йодидом утворює йод:
SO42-+Ba(JO3)2=BaSO4+ 2JO3-,
JO3-+5J- + 6H+ = 3J2 + 3H2O.
Одержаний J2 визначають фотометрично з крохмалем у формі адсорбційної сполуки синього кольору. Цей метод високочутливий. Заважають визначенню HCO3-, SO32-, BO2-.
Mn2+
окиснюють
у перманганат рожево-фіолетового
кольору,
![]() =
2103,
чутливість 0,5 мкг/мл. Як окисники
використовують KJO4,
(NH4)2S2O8,
NaBiO3
у середовищі сульфатної та фосфатної
кислот:
=
2103,
чутливість 0,5 мкг/мл. Як окисники
використовують KJO4,
(NH4)2S2O8,
NaBiO3
у середовищі сульфатної та фосфатної
кислот:
2Mn2+ + 5IO4- + 3H2O = 2MnO4- + 5IO3- + 6H+,
2Mn2+ + 5S2O82- + 8H2O = 2MnO4- + 10SO42- + 16H+.
При окисненні пероксодисульфатом потрібно додавати невелику кількість Ag2SO4 чи солі кобальту, які каталізують реакцію.
Визначенню заважають Cl-, Ce4+, Ni2+, Co2+, Cu2+, Cr2O72-, UO22+.
Використовують взаємодію мангану (ІІ) з формальдоксимом |
|
при
рН 9,5-10,5, у результаті чого утворюється
комплекс, який окиснюється киснем у
червоно-бурий продукт [Mn(CH2NO)6]2-,
![]() =
1,12104.
Визначенню заважають важкі метали
(Cu2+,
Co2+,
Ni2+,
Zn2+,
Cd2+),
які маскують ціанідом.
=
1,12104.
Визначенню заважають важкі метали
(Cu2+,
Co2+,
Ni2+,
Zn2+,
Cd2+),
які маскують ціанідом.
Ті(IV) утворює з гідроген пероксидом, якого потрібний пятикратний надлишок, у середовищі 0,5–1М H2SO4 жовто-оранжевий комплекс:
Ti(OH)22+ + H2O2 = [Ti(OН)2H2O2]2+.
Визначення
не надто чутливе,
![]() =7,0102,
підвищує чутливість присутність у
розчині комплексону III. Заважають
визначенню Fe3+,
окисники, фториди і велика кількість
фосфатів, які утворюють комплекси з Ті.
Визначення тітану неможливе у присутності
ванадію та молібдену.
=7,0102,
підвищує чутливість присутність у
розчині комплексону III. Заважають
визначенню Fe3+,
окисники, фториди і велика кількість
фосфатів, які утворюють комплекси з Ті.
Визначення тітану неможливе у присутності
ванадію та молібдену.
Титан можна визначати фотометрично з хромотроповою кислотою:
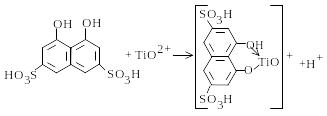
У кислому розчині з рН 2-3 титан з хромотроповою кислотою утворює комплекс червоно-бурого кольору. Заважає визначенню Fe3+, який утворює комплекс темнозеленого кольору.
Fe2+
при рН 2,5-9,0 утворює з 1,10-фенантроліном
оранжево-червоний комплекс (![]() =1,11104)
або з 2,2-дипіридилом
рожевий
(
=1,11104)
або з 2,2-дипіридилом
рожевий
(![]() =8,65104):
=8,65104):
1,10-фенантролін
2,2-дипіриділ |
Комплекс Fe2+ з 1,10-фенантроліном |



Якщо розчин містить сполуки феруму у різних ступенях окиснення, то перед визначенням ферум (ІІІ) відновлюють гідроксиламіном до феруму (ІІ).
Нерозчинні та звязані у комплекси форми феруму необхідно попередньо перевести у розчинний стан. При кипятінні розчину природного об’єкта всі форми сполук фосфору перетворюються у РО43-, який зв’язує ферум. У цьому випадку при великому вмісті фосфатів спочатку відділяють сполуки феруму, а потім їх визначають. Заважають іони Cu2+, Co2+, Ni2+, Zn2+ у концентраціях, що перевищують концентрацію феруму, а Bi3+, Ag+, Hg2+ при вмісті, більшому за 1 мг/л. Ціаніди заважають, але їх видаляють кислотою.
Fe3+ утворює із сульфосаліциловою кислотою різні комплексні сполуки при різних значеннях рН. Комплекси стійкі, тому Fe3+ можна визначити за наявності фосфатів, ацетатів, боратів. У кислому середовищі визначенню не заважають Cu2+ і Al3+, бо їхні сполуки із сульфосаліциловою кислотою менш стійкі та безбарвні. В лужному середовищі визначенню Fe(III) не заважають іони F-.
|
рН 1,8-2,5 |
рН 4-8 |
рН 8-11 |
|---|---|---|---|
|
|
|
|
|
фіолетовий, =1,6103 |
червоно-
бурий,
|
жовтий,
|



Al3+ утворює комплекси з низкою реагентів. Нижче показані комплекси з:
еріохромціаніном R, |
алюміноном, |
|
|
пірокатехіновим фіолетовим, |
алізарином S |
|
|
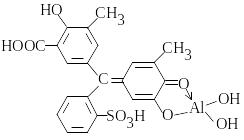

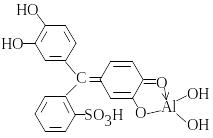
З
еріохромціаніном
R
при рН=5,4 утворює фіолетового кольору
сполуку, яку фотометрують,
![]() =4104.
Заважає визначенню Fe3+,
який відновлюють у Fe2+
дією NH2OH.
=4104.
Заважає визначенню Fe3+,
який відновлюють у Fe2+
дією NH2OH.
З алюміноном (С22H11O9(NH4)3) Al(OH)3 утворює комплекс у вигляді колоїдного розчину яскраво-червоного кольору, =24103:
Комплекс стабілізують желатином, оптимальне рН 3,8-4. З алюміноном можна визначити до 0,05 мг/л алюмінію. Визначенню заважає низка речовин, які усувають таким чином: органічні речовини обзолюють концентрованою H2SO4; сульфіди (понад 10 мг/л) окиснюють при кип’ятінні з гідрогенпероксидом; хлориди (>0,5 мг/л) випарюють насухо в присутності H2SO4; фосфати розчиняють кислотою; сполуки феруму маскують цитратною та тіогліколевою кислотою.
З пірокатехіновим фіолетовим при рН 6,1 Al3+ утворює сполуку голубого кольору. Межа виявлення становить 0,001 мкг/мл. Цей метод застосовують для визначення вмісту алюмінію у поверхневих, ґрунтових та морських водах, які містять розчинений алюміній.
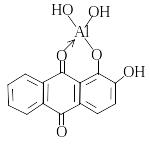
З алізарином S (1,2-діоксиантрахіноном) при рН 4,8, при якому сам алізарин мАє жовтий колір, у розчинному стані та в стані гідроксоформ утворюється адсорбційна сполука червоно-фіолетового кольору.
Екстракційно-спектрофотометричні методи
Спектрофотометрію, УФ та видиму, часто комбінують з екстракцією. Так визначають низку мікроелементів у водах та ґрунтових витяжках з одночасним їх концентруванням та відділенням від інших мікроелементів.
Ni2+
визначають
з
диметилгліоксимом
в аміачному середовищі. Нерозчинна у
воді внутрішньо-комплексна сполука
рожевого кольору екстрагується неполярним
розчинником (CHCl3,
CCl4)
у формі жовтого комплексу,
![]() =1,3104:
=1,3104:
|
|
|


Заважають визначенню Al3+, Mo, Cu2+, Fe3+, Mn2+. Нікель можна визначати у присутності Co2+, V.
Визначення
можна провести без
застосування екстракції,
якщо у розчин ввести окисник: H2S2O7,
галогени (переважно J2),
а також, як описано в роботі (Зінчук
В.К., Вовк І., 2008) H2SO5.
Утворюється сполука коричневого кольору,
для якої при рН 10,2 (0,03 М Na4P2O7)
=12,4103,
а при рН 8,3-8,6 (0,15 М амонійна буферна
суміш)
![]() =12103.
Межі визначуваних концентрацій –
0,05-0,90 мг/л. Інші важкі метали визначенню
не заважають.
=12103.
Межі визначуваних концентрацій –
0,05-0,90 мг/л. Інші важкі метали визначенню
не заважають.
Со2+
з нітрозо-R-сіллю
жовтого кольору в кислому середовищі
утворює комплекс червоного кольору;
![]() =3,5104,
=3,5104,
![]() =1,5104.
=1,5104.
|
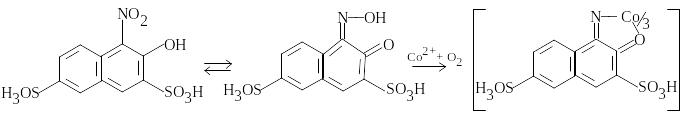
Заважають визначенню Cu2+, Ni2+, Fe2+, Fe3+, Mn2+, тому Со2+ попередньо від них відділяють.
У
концентрованому розчині тіоціанату
при рН 3-5 Со2+
утворює голубий комплекс [Co(SCN)4]2-,
який швидко руйнується. Комплекс
екстрагують органічними розчинниками
і визначають: при екстракції ізоаміловим
спиртом –
![]() =1103,
циклогексаном –
=2104.
Заважають визначенню Fe3+,
V, Bi3+,
UO22+,
Mo, W, Cu2+,
Ag+
Ni2+,
Hg2+,
Cr2O72-.
=1103,
циклогексаном –
=2104.
Заважають визначенню Fe3+,
V, Bi3+,
UO22+,
Mo, W, Cu2+,
Ag+
Ni2+,
Hg2+,
Cr2O72-.
З
-нітрозо--нафтолом
в
аміачному середовищі Со2+
утворює сполуку, яку екстрагують
толуолом. Заважають визначенню іони
важких металів. Спочатку сполуки феруму
окиснюють гідрогенпероксидом до феруму
(ІІІ) і зв’язують заважаючі іони Fe3+,
Al3+,
Ca2+,
Mg2+
цитратом. Після завершення екстракції
комплексу з кобальтом відділяють іони
більшості важких металів реекстракцією
HCl (комплекси в кислому середовищі
руйнуються). В лужному середовищі
фотометрично визначають утворений
комплекс кобальту,
![]() =2,6104:
=2,6104:

Cd2+ визначають екстракційно-фотометрично в лужному середовищі з дитизоном після відділення всіх інших іонів. Внаслідок взаємодії кадмію з дитизоном утворюється первинний дитизонат (HDz)2Cd, досить погано розчинний у карбон тетрахлориді та добре розчинний в хлороформі, =8,8104.

Іони Ni2+, Co2+ маскують додаванням диметилглiоксиму, а iони iнших металів (Сu2+, Ag+) потрібно попередньо екстрагувати дитизоном у кислому середовищі (pH 2). Заважає визначенню Zn2+, якщо його вміст у 500 разів більший від вмісту кадмію, а також окисники - хлор, бром, пероксиди. Не заважають визначенню хром, алюміній, ферум, манган, фосфати, тіоціанати, сульфати, сульфіди.
Zn2+
визначають екстракційно-фотометрично
з дитизоном
у хлороформі
при рН 4,0-5,5, а для усунення впливу інших
важких металів (Cu2+,
Cd2+,
Pb2+,
Ni2+,
Co2+,
Hg2+,
Sn2+,
Bi3+)
їх зв’язують тіосульфатом, і лише за
умови 100-кратного перевищення концентрації
Zn2+
для маскування застосовують ціанід.
Заважають визначенню лише Sn та Fe3+,
останній осаджують у формі Fe(OH)3.
Оскільки тіосульфат є відновником, то
недопустима присутність у розчині
окисників, які усувають кип’ятінням.
Аналогічно, як і у випадку визначення
кадмію, утворюється забарвлений комплекс,
який екстрагують CCl4.
Метод чутливий,
![]() =9,4104.
=9,4104.
Pb2+ визначають теж з дитизоном у вигляді червоного комплексу при рН8-8,5, який екстрагують дією CСl4 або CHCl3, =6,9104. Оскільки більшість важких металів утворюють аналогічні комплекси, то визначення можливе після попереднього відділення та маскування катіонів інших металів. Тому спочатку при рН 2 екстрагують комплекс [Cu(HDt)2]. Слід зазначити, що при цьому можна кількісно визначити вміст Cu2+. Після усунення купруму доводять рН до 7, додають K4[Fe(CN)6] для зв’язування цинку в Zn2[Fe(CN)6]. Для усунення впливу іонів мангану, феруму до водного розчину додають гідроксиламін, цитрат або тартрат, доводять рН до 8-8,5 і екстрагують комплекс [Pb(HDt)2] органічним розчинником.
Cu2+ утворює малорозчинний комплекс з діетилдитіокарбаматом, який екстрагують карбон тетрахлоридом
Для
комплексу в розчині СCl4
![]() =1,4104,
чутливість визначення 0,1 мкг/мл. Щоб
усунути вплив інших катіонів, які
утворюють забарвлені комплекси з
діетилдитіокарбаматом, спочатку
одержують комплекс з плюмбумом, з якого
Cu2+
селективно витискає Pb2+
з утворенням ще стійкішого комплексу:
=1,4104,
чутливість визначення 0,1 мкг/мл. Щоб
усунути вплив інших катіонів, які
утворюють забарвлені комплекси з
діетилдитіокарбаматом, спочатку
одержують комплекс з плюмбумом, з якого
Cu2+
селективно витискає Pb2+
з утворенням ще стійкішого комплексу:


У цьому випадку можуть заважати лише іони Ag+, Hg2+, Ti3+, Bi3+.
V
визначають з 8-гідроксихіноліном
при рН 4.
Утворюється
жовто-бурого кольору комплекс, який
екстрагують хлороформом,
![]() =3,3103.
Органічні речовини у розчині попередньо
окиснюють дією HNO3
або руйнують сплавлянням з лугом.
Заважають іони алюмінію, феруму, бісмуту,
тітану. Їх вплив усувають повторною
реекстракцією буферним розчином з
хлороформного екстракту. Визначенню
заважає лише велика кількість W.
=3,3103.
Органічні речовини у розчині попередньо
окиснюють дією HNO3
або руйнують сплавлянням з лугом.
Заважають іони алюмінію, феруму, бісмуту,
тітану. Їх вплив усувають повторною
реекстракцією буферним розчином з
хлороформного екстракту. Визначенню
заважає лише велика кількість W.
Мо визначають у формі комплексу з тіоціанатом після відновлення Мо (VI) до Мо (V). Як відновник застосовують SnCl2. Комплекс утворюється при концентрації тіоціанату 0,3 М, а з вольфрамом – при концентрації 0,6 М, отже сполуки вольфраму не заважають. Не заважають також сполуки ванадію, які відновлюються відновником. Сполуки хрому видаляють у формі хлориду при випаровуванні. Заважає визначенню Fe3+, його відновлюють до Fe2+ дією надлишку SnCl2. Щоб Мо(VI) відновився до Mo(V), а не до нижчих ступенів окиснення, залишок SnCl2 руйнують додаванням нітриту. Одержаний комплекс екстрагують ізоаміловим спиртом у кислому середовищі, =15,3103. Недоліком визначення є можливість відновлення сполук молібдену до нижчих ступенів окиснення, що призводить до пониження точності результатів аналізу.