

9.4. Загальні кількісні характеристики, спільні для ґрунтів та вод
Визначення кислотності ґрунту
Активна кислотність зумовлена вмістом вільних іонів H3O+ у ґрунтовому розчині і діє безпосередньо на кореневу систему рослин та ґрунтові мікроорганізми. Активну кислотність визначають з метою впливу на ґрунт добрив, підбору культур для сівозміни.
Одержують водну витяжку з ґрунту при співвідношенні маси ґрунту (г) до об’єму води (мл) як 1:2,5. В одержаному фільтраті pH-метром вимірюють значення pH.
Потенційна кислотність зумовлена наявністю іонів H3O+ та Al3+ у ГПК, здатних обмінюватись на інші іони. Потенційна кислотність поділяється на обмінну та гідролітичну.
Обмінну кислотність визначають у процесі взаємодії ґрунтового поглинального комплексу з нейтральними солями – розчином 1М KCl. У цьому випадку з ГПК можуть витіснятися легкозв’язані іони гідрогену, а також іони алюмінію, які у водному розчині підкислюють середовище внаслідок гідролізу:
[ГПК](H+,Al3+)
+ 4KCl
![]() [ГПК](4K+)
+ HCl + AlCl3,
[ГПК](4K+)
+ HCl + AlCl3,
AlCl3
+ H2O
![]() AlOHCl2
+ HCl.
AlOHCl2
+ HCl.
Вміст кислоти визначають титриметрично. Для цього одержану ґрунтову витяжку титрують стандартизованим 0,0200 М розчином лугу з індикатором метиловим червоним. Обчислюють обмінну кислотність:
![]()
![]() ,
,
де g – наважка повітряно-сухого ґрунту, г; К коефіцієнт вологості.
Можна визначити кислотність, зумовлену лише іонами H3O+ (спрощено Н+). Для цього, після одержання витяжки, перед титруванням зв’язують іони Al3+ натрій фторидом з одержанням Na3[AlF6], після чого розчином лугу відтитровують кислоту, виділену з ґрунтового поглинального комплексу за реакцією:
[ГПК](H+, Al3+) + KCl + 6NaF [ГПК](K+, 3Na+) + HCl + Na3[AlF6].
За різницею обох титрувань обчислюють вміст іонів Al3+, витиснених з ґрунту. Так обчислюють Hобм.(H+,Al3+) та Hобм.(H+).
За одержаними даними визначають вміст алюмінію:
m(Al3+) = [Hобм.(H+,Al3+) – Hобм.(H+)]M(Al3+), мг/100 г.
Обмінну кислотність, обумовлену як кислотами, так і солями алюмінію, можна визначити і потенціометрично у витяжці 1 М KCl.
Гідролітична кислотність зумовлена іонами гідроксонію, які міцніше зв’язані в ГПК і можуть витіснятися лише дією розчину з лужною реакцією середовища (0,1 М CH3COONa, NaOH):
[ГПК](H+) + СH3COONa [ГПК](Na+) + СH3COOH,
[ГПК](Al3+)+3СH3COONa +3H2O [ГПК](3Na+) +3СH3COOH+Al(OH)3.
Виділену кислоту у фільтраті відтитровують стандартним 0,0200 М розчином лугу за наявності фенолфталеїну. Обчислюють гідролітичну кислотність:
![]()
За гідролітичною кислотністю обчислюють кількість кальцій карбонату, необхідного для вапнування кислого ґрунту. Так, для нейтралізації 1 ммоль H3О+ на 100 г ґрунту треба внести 50 мг CaCO3. Маса орного шару ґрунту товщиною до 20 см на 1 га становить 3000 т. Тоді маса CaCO3, потрібного для нейтралiзацiї кислотності, становить 50 Hгідр. мг/100 г. Звідси:
 .
.
Отже, на нейтралізацію 100 м2 ґрунту з обчисленою гідролітичною кислотністю Hгідр. витрачається маса СaCO3:
m(CaCO3) = 15 Hгідр. (кг).
Визначення кислотності та лужності води
Під кислотністю води розуміють вміст у воді всіх речовин, які здатні вступати у взаємодію з розчинами лугів.
Активну кислотність, як величину рН, визначають потенціометрично. Однак, активна кислотність не відображає, як різні сполуки здатні підкислювати воду: сильні кислоти; слабкі кислоти, такі як ацетатна, сульфітна, карбонатна, сульфідна; катіони, які внаслідок гідролізу підкислюють воду – NH4+, Fe3+, Fe2+, Al3+, катіони органічних основ. Тому окремо визначають ці групи сполук, титруючи однакові порції води 0,100 М стандартним розчином лугу з різними індикаторами.
Загальну кислотність визначають, титруючи порцію води (переважно 100,0 мл) робочим розчином лугу з індикатором тимолфталеїном (рТ 10) або фенолфталеїном (рТ 8,9). Можна використати потенціометричне титрування:
HAn + OH- An- + H2O, де HAn – слабка кислота,
H3O+ + OH- 2H2O.
Сильні кислоти титрують розчином лугу з індикатором метиловим червоним (рТ 5):
H3O+ + OH 2H2O,
H3PO4 + OH- H2PO4- + H2O.
Вміст слабких кислот (органічних, карбонатної та сульфідної, Н2РО4-) та катіонів слабких основ обчислюють як різницю двох попередніх титрувань.
Лужність води зумовлена всіма речовинами, які вступають у взаємодію з іонами гідроксонію. Сюди відносяться сильні основи, слабкі основи, такі як аміак, анілін, піридин; аніони слабких кислот, які внаслідок гідролізу підлужнюють воду – HCO3-, CO32-, H2PO4-, HPO42-, HSO3-, SO32-, HS-, S2-.
Загальна лужність, або вміст іонів ОН-, НСО3- та СО32-, визначається титруванням проби води 0,100 М розчином сильної кислоти (стандартизованої за Na3B4O7) до рН 3,5 з індикатором метиловим оранжевим:
ОН- +Н3О+ 2Н2О,
НСО3- + Н3О+ Н2СО3+ Н2О,
СО32- + 2Н3О+ Н2СО3+ 2Н2О.
Якщо титрування проводити потенціометрично, тоді на кривій титрування можна розрізнити дві точки перегину.
Вільна лужність обумовлена вмістом іонів ОН- та СО32-, а також домішками S2-, PO43-, SiO32-. Її визначають титруванням проби води розчином HCl до рН 8,9 за фенолфталеїном.
Слабкі основи, такі як НСО3-, НPO42-, НS-, аніони гумінових кислот, визначають як різницю між попередніми титруваннями.
Визначення лужності води можна проводити після попередньої відгонки при тривалому кип’ятінні летких основ (аміаку, амінів). У залишку води після відгонки, за умови попередження доступу СО2 з повітря, проводять титрування основ розчином кислоти. У цьому випадку можна визначити окремо вміст летких основ.
Якщо стічна вода має високий вміст гіпохлориту, його попередньо відновлюють, оскільки гіпохлорит руйнує індикатори, які застосовують для титрування.
Визначення карбону органічних речовин
Карбон у воді входить у склад розчинних гідрогенкарбонатів кальцію та магнію, які зумовлюють карбонатну твердість води, у склад водорозчинних органічних речовин та комплексів важких металів з гуміновими кислотами (переважно фульвокислотами).
У ґрунті карбон є в складі важкорозчинних карбонатів, у ґрунтовому розчині у вигляді гідрогенкарбонатів, подібно як і в воді, у складі органічних сполук, у складі специфічних гумусових речовин. Для того, щоб визначити вміст карбону органічних сполук, необхідно виключити карбон карбонатів.
Вміст карбону зручно визначати за кількістю виділеного СО2. Описані різні методики визначення карбону органічних речовин. Визначають загальний вміст СО2, утвореного окисненням органічних речовин та розкладом карбонатів, та кількість СО2, утвореного лише під час розкладу карбонатів.
Одну пробу ґрунту прожарюють при 1000С у присутності каталізатора СоО. При цьому органічні сполуки окиснюються до СО2, H2O, N2, а карбонати розкладаються до СО2. Вміст виділеного СО2 визначають за характеристичними коливаннями молекули методом інфрачервоної спектроскопії.
У випадку аналізу води, пробу води попередньо випаровують, потім процедуру з сухим залишком продовжують аналогічно.
Другу таку саму пробу води або ґрунту при 150С обробляють розчином HCl для розкладу неорганічних карбонатів, а за виділеним СО2 визначають вміст карбону карбонатів.
За різницею загальної кількості виділеного СО2 та виділеного внаслідок розкладу карбонатів вираховують кількість СО2, утвореного окисненням органічних речовин, а за кількістю СО2 обчислюють кількість карбону, що припадає на органічні речовини.
Для визначення СО2, одержаного окисненням органічних речовин до СО2 використовують спеціально сконструйовану установку (рис.9.1).
Рис.9.1. Пристрій для визначення карбону органічних речовин у воді |
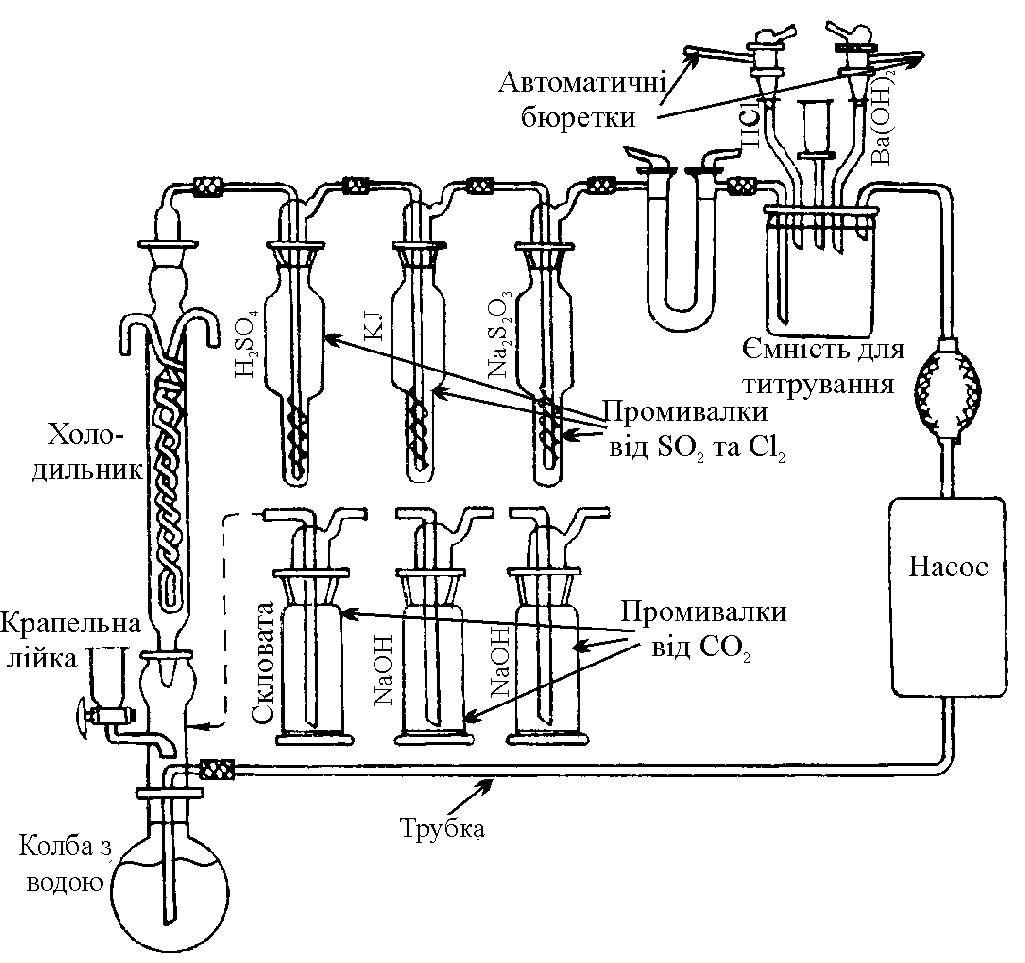
При вмісті у воді карбону органічних речовин у межах 3-400 мг/л як окисник застосовують суміш CrO3 і P2O5 у присутності каталізатора Ag2SO4. Оскільки визначенню заважають хлориди, сульфіти, то хлориди окиснюють, діють кислотою, а утворені Cl2, SO2 та CO2 вловлюють.
Спочатку пробу води (25,0 мл) у колбі з доданими (через крапельну лійку) декількома мілілітрів концентрованої H2SO4 нагрівають протягом 15 хв при 40-60С, під’єднавши насос через промивалки з лугом для усунення СО2. При цьому видаляється СО2, який був у повітрі та який утворився під час взаємодії карбонатів, які були у воді, з кислотою. Виділений СО2 вловлюють 0,1 М розчином лугу, а наявні SO2 та Cl2 – розчинами відновників у інших промивалках.
Після видалення з води СО2, який утворився при розкладі карбонатів та з повітря, зупиняють насос.
У посудину для титрування вливають стандартизований 0,100 М розчин Ba(OH)2.
У колбу через лійку вводять суспензію суміші окисника, концентровану сульфатну кислоту, Ag2SO4 і все разом кип’ятять протягом 60 хв. Відбувається окиснення органічних речовин, які є у воді, з виділенням СО2. Колбу під’єднують до наноса трубкою і знову включають насос. Виділений у процесі окиснення органічних речовин СО2 вловлюється титрованим розчином Ba(OH)2:
Ва(ОН)2 + 2СО2 Ва(НСО3)2.
Залишок Ва(ОН)2 відтитровують стандартизованим розчином HCl з індикатором фенолфталеїном, при цьому НСО3- не титрується:
Ва(ОН)2+2НCl BаCl2 + 2H2O.
Обов’язково проводять сліпу пробу.
Обчислюють вміст карбону органічних речовин:
![]() ,
,
де V(HCl) – об’єм розчину, затраченого на титрування проби та сліпої проби, мл; СЕ та МЕ – відповідно молярна концентрація еквівалента, моль/л, та молярна маса еквівалента, г/моль.
Якщо попередньо було визначено загальний вміст органічних речовин у воді, то для вод зі сталим постійним вмістом органічних речовин вираховують показник забруднення води:
![]() ,
,
який залежно від типу стоку набуває різних значень.
Стічні води, залежно від переважаючого забруднення окремими класами органічних речовин, мають такі значення коефіцієнта К:
побутові стічні води та стоки харчової промисловості – К=2,4 -2,5;
стоки з переважанням насичених вуглеводнів – К=1,20-1,25;
стоки з переважанням ненасичених вуглеводнів – К=1,17;
стоки з ароматичними вуглеводнями – К<1,10.
Використовуючи величину коефіцієнта, можна орієнтовно оцінити, які органічні речовини переважно забруднюють воду.
Визначення загального вмісту сполук нітрогену у ґрунті
Загальний вміст нітрогену в ґрунті залежить від вмісту гумусу і коливається в межах 0,01-0,03% (піщані підзолисті), 0,4-0,6% (чорноземи), 3-4% (торф’яні ґрунти). У ґрунті є три групи сполук нітрогену, в різній мірі доступні рослинам:
водорозчинні мінеральні сполуки (NO2-, NO3-, NH4+, NH3) з вмістом декілька мг/кг сухого ґрунту. Їх витісняє вода або розчини нейтральних солей. Це легкозасвоювані рослинами форми;
легкогідролізовані органічні сполуки нітрогену (десятки мг/кг ґрунту). Їх переводять у розчинний стан дією 0,25 М H2SO4. Ці форми сполук нітрогену рослини можуть засвоювати;
важкогідролізовані органічні сполуки нітрогену (основна частина валового вмісту нітрогену в ґрунті). Їх можна перевести в розчинний стан дією концентрованої мінеральної кислоти.
У гумусовому шарі ґрунту переважає нітроген у складі органічних сполук, а на мінеральні форми (NH4+, NO2-, NO3-) припадає 1-3%. У нижніх шарах ґрунту міститься фіксований NH4+, на який припадає 30-60% всього нітрогену.
Визначають вміст нітрогену за методом К’єльдаля.
Пробу ґрунту обзолюють кип’ятінням з концентрованою H2SO4 у присутності як каталізатора Se. При цьому визначають і органічний, і амонійний нітроген.
Відбуваються такі реакції за участю селену як каталізатора:
Se +2H2SO4 = H2SeO3 +2SO2 +H2O.
Одержана селенітна кислота руйнує органічні речовини:
або
CxHyOzNH2
+H2SO4![]() CO2+H2O+NH2SO2OH,
CO2+H2O+NH2SO2OH,
а дальше утворюються солі амонію:
NH2SO2OH+ H2SO4 NH4HS2O7,
NH4HS2O7+H2O![]() NH4HSO4+H2SO4.
NH4HSO4+H2SO4.
Одержаний після окиснення розчин охолоджують, переносять у колби для відгонки. Дистиляційні колби (рис.9.2) через холодильник сполучають з колбами для вловлювання аміаку, додають у дистиляційні колби розчин лугу і нагрівають суміш до повного видалення NH3:
NH4HSO4 + 2NaOH Na2SO4 + NH3 +2H2O.
На кожні 10 мл кислоти для обзолення припадає 40 мл 33% розчину лугу.
Рис.9.2. Пристрій К’єльдаля для дистиляції аміаку |
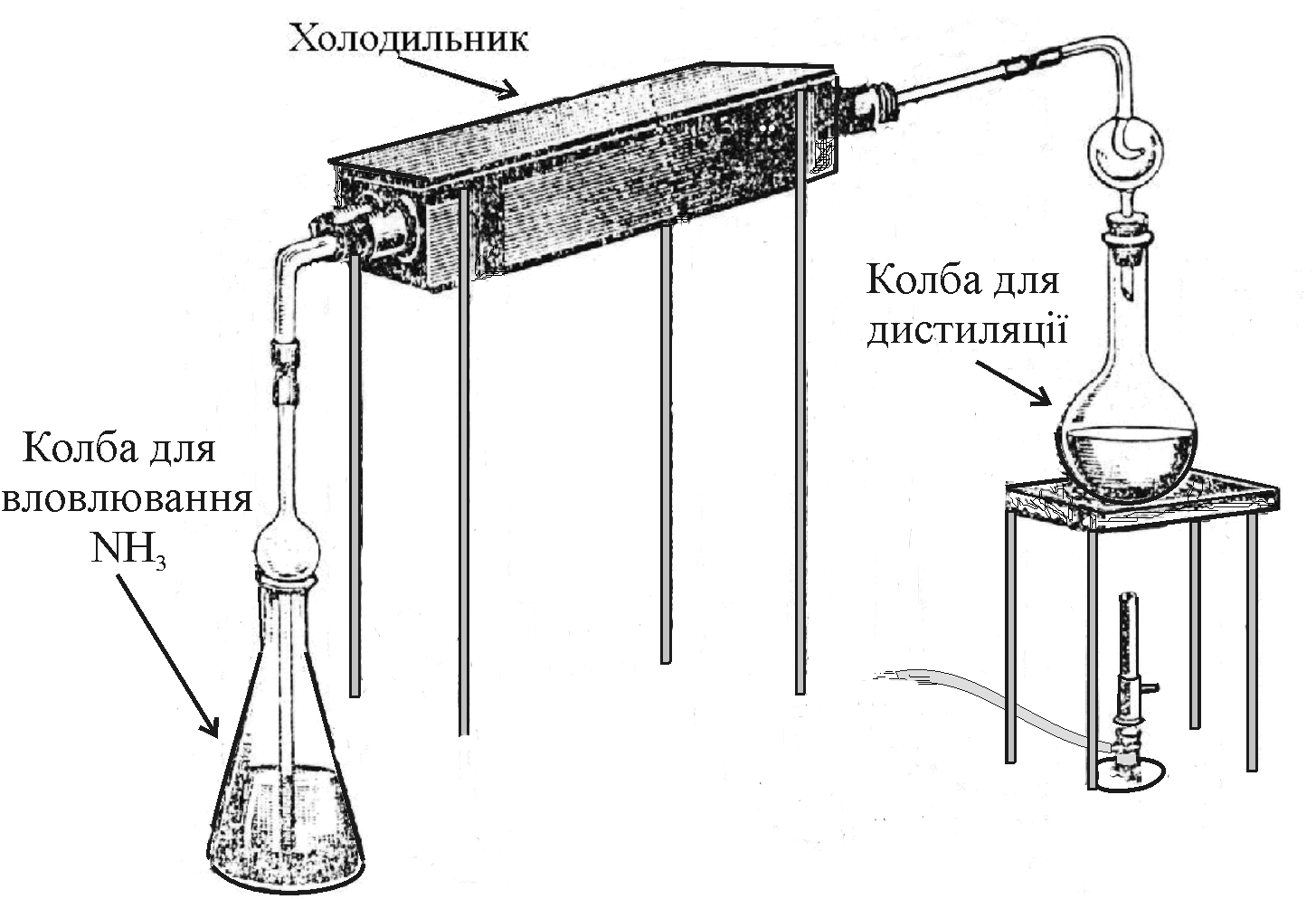
Процес відгонки припиняють, коли у колбі залишається третина від початкового об’єму розчину. Аміак вловлюють розчином кислоти. Якщо для вловлювання використовують стандартизований розчин H2SO4, то надлишок кислоти відтитровують стандартизованим розчином лугу, а якщо вловлюють аміак розчином Н3ВО3, то утворений (NH4)3BO3 титрують стандартизованим розчином H2SO4. Можна також визначати амоній фотометрично з реактивом Неслера.
Для визначення вмісту всіх форм нітрогену в різних ступенях окиснення (-3, +3 до +5) обзолення ведуть фенолсульфатною кислотою:
о-OH-C6H4-SO2OH+HONO2 = о-OH-C6H4-NO2+H2SO4,
а утворений нітрофенол відновлюють до амінофенолу цинком:
о-OH-C6H4-NO2+Zn+4H+ = о-OH-C6H4-NH2+2H2O.
Далі визначення проводять аналогічно, як описано вище.
Визначення нітрогену органічних сполук у воді. Пробу води випаровують насухо, потім прожарюють у присутності окисника. Для окиснення беруть таку кількість води, щоб у ній було 2-6 мг нітрогену. Якщо стічна вода містить багато нітритів та нітратів, то перед окисненням до неї додають розчин FeSO4, який відновлює ці сполуки нітрогену до оксидів, останні видаляються і не заважають визначенню.
Окиснення проводять сумішшю концентрованої H2SO4 та K2SO4 при 315-370C з каталізатором Cu2+ (можна Hg2+) до утворення (NH4)2SO4, з якого лугом видаляють NH3 і визначають. Окремо визначають вміст NH4+ у воді. За різницею вираховують вміст нітрогену, який припадає на органічні сполуки. Відганяють аміак аналогічно, як описано вище. За таких умов окиснення не розкладаються гетероциклічні нітрогеновмісні речовини, азиди, азосполуки, нітро-, нітрозосполуки, оксими.
Визначення легкогідролізованого нітрогену органічних сполук у ґрунті. Одержують ґрунтову витяжку дією 0,5 М H2SO4. При необхідності у витяжці руйнують органічні сполуки дією Н2О2. У витяжку потрапляють як неорганічні сполуки нітрогену, так водорозчинні та гідролізовані форми органічних сполук. Визначення виконують фотометрично, вимірюючи оптичну густину комплексної сполуки червоно-бурого кольору, яка утворилася внаслідок дії реактиву Неслера на аміак:
NH3 + OH- + 2 [HgJ4]2- [J2Hg2NH2]J + 5J-+ H2O.
Для усунення впливу катіонів металів, які заважають, перед додаванням реактиву Неслера в розчин вводять сегнетову сіль KNaC4H4O64H2O.
Якщо
концентрація продукту дуже мала, то
сполука перебуває у стійкому дисперсному
стані, особливо при додаванні захисного
колоїду: желатину, полівінілового
спирту, гуміарабіку. Максимум поглинання
припадає на =400 нм,
молярний коефіцієнт
![]() =5,0103,
чутливість визначення 0,07 мкг/мл.
=5,0103,
чутливість визначення 0,07 мкг/мл.
Дослідженням (Демуцкая Л.Н., Калиниченко И.Е., 1999) встановлено, що використання готового препарату реактива Неслера може давати занижені результати щодо вмісту NH4+. Найкраще готувати реактив безпосередньо перед визначенням NH4+, так щоб концентрації компонентів становили 0,220 М Hg2+, 0,885 M J-, 4 M NaOH.
Так визначають лише амоній та амоній, утворений з легкогідролізованих органічних речовин.
Потім у такій же ґрунтовій витяжці при повільному кип’ятінні відновлюють усі форми нітрогену порошком цинку до амонію. Після охолодження додають концентровану H2SO4 і випаровують розчин доти, поки почнуть виділятися пари SO2 та залишок побуріє. Залишок розчиняють в 1 М KCl. В одержаному розчині аналогічно визначають вміст NH4+.
Визначення амонію у ґрунті та воді
Амоній, який є в ґрунті у вигляді водорозчинних солей та в обмінній формі, переводять у розчин сольовою витяжкою: 1 М KCl, 0,05 М NaCl або 0,05 М K2SO4. Для зв’язування iонiв Ca2+ та Mg2+ до розчину додають сегнетову сiль. Визначення проводять фотометрично з реактивом Неслера або у формі індофенольної сполуки.
Аміак
у лужному середовищі при взаємодії з
гiпохлоритом
та фенолом
утворює сполуку синього кольору. Ця
реакція лежить в основі селективного
та чутливого фотометричного визначення
нітрогену (![]() =
4,5103,
чутливість 0,07 мкг/мл).
=
4,5103,
чутливість 0,07 мкг/мл).

Визначення амонію у воді. У забрудненій воді безпосередньо з проби визначають амоній різними способами.
За умови великої кількості амонію за методом К’єльдаля дією лугу відганяють NH3 з наступним вловлюванням розчином Н3ВО3.
Визначенню заважають інші леткі сполуки. Щоб цьому перешкодити, поступають так:
аміни і піридин попередньо визначають у воді;
хлораміни, утворені хлоруванням води, відновлюють дією S2O32-, AsO2-;
феноли зв’язують у феноляти, створивши більш лужне середовище.
Якщо кількість амонію мала, його відразу визначають фотометрично.
Зручно визначати NH4+ методом прямої потенціометрії.
Визначення нітрату. Нітрат у ґрунті є в рухливій формі. У розчин його переводять або витяжкою 0,05 М K2SO4, або водою.
У одній частині витяжки визначають вміст NH4+ фотометрично з реактивом Неслера, а в іншій порошком Zn відновлюють NO3- до NH4+ і також визначають вміст амонію. За різницею вираховують вміст NO3-.
У
водній витяжці нітрат визначають
фотометрично
з дисульфофеноловою
кислотою
у формі тринітрофеноляту калію
жовто-оранжевого кольору, для якого
![]() =9,4103,
чутливість 0,1 мкг/мл:
=9,4103,
чутливість 0,1 мкг/мл:
|

Визначенню заважає значна кількість хлориду та нітриту в розчині. Іони амонію попередньо випарюють з лужного розчину.
У воді нітрат можна визначати фотометрично з натрій саліцилатом у формі жовтого нітросаліцилату в межах концентрацій 0,1-20 мг/л:

Визначенню заважають забарвлені органічні речовини, Fe3+, які усувають співосадженням на Al(OH)3. Іони Cl- (>200 мг/л) зв’язують, додаючи Ag2SO4. У випадку надміру нітриту у воді до неї додають амоній сульфат і кип’ятять пробу до видалення N2:
NO2- + NH4+ N2 + 2H2O.
Фотометрично у формі NH3 з реактивом Неслера визначають NO3- після відновлення до NH3 порошком алюмінію.
Якщо вміст нітрату малий, то кип’ятінням видаляють NH3, а потім відновлюють нітрат до амонію дією Al. Якщо вміст нітрату великий, то великий і вміст NO2-. Тому попередньо до води додають NH4Cl, а утворений NH4NO2 при випаровуванні розкладається до N2.
Безреагентне визначення нітрату у водах можна проводити за поглинаннм при =436 нм згідно стандарту DIN 38404-C3 (див. Хоффманн М., Галаган А.А., 1999). Розроблено низку методик для визначення вмісту нітрату у природних водах за поглинанням іоном NO3- в УФ діапазоні випромінювання без додавання реагентів. Максимум поглинання NO3- припадає на 210 нм, а наявні у воді гумати та нітрит поглинають при 220 нм, йодид – 230 нм.
З ґрунтової витяжки у 0,05 М K2SO4 можна визначати нітрат потенціометрично з іоноселективним електродом.
Визначення фториду
Надлишкова кількість фториду у ґрунті та воді свідчить про забруднення. У ґрунті він зв’язаний у важкорозчинний CaF2.
Пробу ґрунту сплавляють з Na2CO3, K2CO3 (4 год при 800С):
CaF2+Na2CO3 CaCO3+2NaF,
після чого додають гарячу воду для розчинення натрій фториду. Після цього додають розчин (NH4)2CO3 для осадження Fe(OH)3. Відфільтровують осад, у розчині виявляють F-.
У пробі води фторид визначають без спеціальної підготовки.
Фотометрично визначають фторид з алізарин-комплексоном при рН 4,6. При цьому La(Ce)-алізарин-комплекс червоного кольору перетворюється у F-La(Ce)-алізарин-комплекс бузково-синього кольору, який фотометрують при λ=610-620 нм. Чутливість визначення – 150 мкг/мл. Заважають визначенню борати, іони Al3+, Fe3+, які утворюють з фторидом стійкі комплекси.
Фотометрично з Zr-алізарин-сульфонатом оранжевого кольору (λ=520-550 нм) визначають фторид як реагент, який утворює комплекс [ZrF6]2- і тим самим руйнується Zr-алізарин-сульфонат – послаблюється його забарвлення. Заважають визначенню окисні сполуки хлору, які попередньо відновлюють дією NaAsO2; Al, Fe.
Потенціометрично з іоноселективним електродом визначають фторид у присутності 1000-кратного надлишку Cl-, Br--, NO3-, SO42-. Заважають лише іони OH- та SiO32-. Так визначають вміст фториду в межах 0,1-110-5 М. Для підтримання сталої іонної сили використовують розчин KNO3 або ацетатний буферний розчин.
Відганяють SiF4 дією на фторид SiO2 при 135С у присутності H2SO4 та Ag2SO4. У відгоні визначають SiF4.
Визначення Fe2+, Fe3+, Al3+ у воді проводять у стічних водах, збагачених цими елементами. При рН 1-1,5 Fe3+ взаємодіє з ЕДТА з утворення стійкого комплексу. Його визначають у пробі води титриметрично з сульфосаліциловою кислотою як індикатором.
Після цього у такій самій пробі води окиснюють Fe2+ у Fe3+ і виконують визначення Fe3+. За різницею обох титрувань вираховують вміст Fe2+.
З такої ж третьої проби води після окиснення відділяють Fe3+ і Al3+ осадженням у формі гідрооксидів амонійним буфером. Осад гідрооксидів Fe(OH)3 та Al(OH)3 відфільтровують і розчиняють в кислоті. При рН 3,5-4,5 на розчин, що містить сполуки феруму (ІІІ) та алюміній, діють точно відомим надлишком стандартного розчину ЕДТА і витримують, бо Al3+ взаємодіє з ЕДТА повільно. Залишок ЕДТА відтитровують стандартизованим розчином ZnCl2. Так визначають суму феруму (ІІІ) та алюмінію. За різницею сумарного вмісту Fe3+ і Al3+ та попередньо визначеного загального вмісту Fe3+ вираховують вміст Al3+.
Визначення рухомої H2SiO3 та оксидів Al2O3, TiO2, Fe2O3 у ґрунті. У витяжці дією 5% КОН з проби ґрунту одержують розчинні силікати та алюмінати в результаті реакцій:
SiO2+2KOH=K2SiO3+H2O,
Al2O3+2KOH=2KАlO2+H2O,
TiO2+2KOH=K2TiO3+H2O,
Fe2O3+2KOH=2KFeO2+H2O.
Після фільтрування та підкислення витяжки розчин містить Al3+, TiO2+, Fe3+, SiO32-. Випаровують розчин насухо в присутності концентрованої HNO3 та декількох крапель Н2О2 (для руйнування залишку органічних речовин), і, таким чином, осаджують нерозчинну H2SiO3. Залишок розчиняють у кислоті. Одержаний розчин містить солі Al3+, TiO2+, Fe3+, в якому комплексонометричним титруванням можна визначити вміст Al3+, Fe3+, а фотометричним методом – вміст титану.