

УЧЕБНИК ВНУТРЕННИЕ БОЛЕЗНИ
.pdfопухолевый клон, способный к дальнейшей пролиферации и распространению.
Данные кариологических исследований свидетельствуют о геномной нестабильности, проявляющейся количественными и структурными изменениями хромосом. Наиболее характерными количественными аномалиями кариотипа при ММ являются моносомия хромосомы 13 и трисомия хромосом 3, 5, 7, 9, 11, 15 и 19.
Все генетические события, определяющие возникновение этой опухоли и ее прогрессирование, проходят несколько этапов, которые условно можно разделить на две большие группы — ранние и поздние.
К ранним, или первичным, событиям онкогенеза можно отнести транслокации с вовлечением локуса 14q32 (трансформация во множественную миелому связана с такими дополнительными хромосомными аномалиями, как делеция длинного плеча хромосомы 13). Дальнейшее распространение опухолево го клона обеспечивается микроокружением костного мозга посредством самоподдерживающего механизма взаимодействия миеломных клеток и костно-мозговых стромальных клеток. На этой фазе болезни миеломные клетки зависимы от ростовых факторов и остаются в костном мозге.
К поздним генетическим и молекулярным событиям относятся хромосомные аберрации с вовлечением 8q24 (локус гена c-MYC), мутации в протоонкогенах N- и K-RAS, а также мутация ТР53. Эти изменения приводят к независимой от стромы костного мозга пролиферации плазматических клеток с последующим переходом в терми нальную фазу болезни и развитием экстрамедулляр ных проявлений.
Важную роль в процессе роста опухоли играют цитокины,
секретируемые миеломными клетками и стромальными элементами костного мозга: ИЛ-6, ИЛ-8, ФНО-α, ИНФ-γ, ИЛ-4.
Изучается роль синдекана – 1 (CD138) в патогенезе ММ.При ММ выявляется мутация гена-супрессора опухолевого роста р53. Большое значение придается опухолевому ангиогенезу. Миеломные клетки синтезируют факторы роста эндотелия сосудов ( VEGF – vascularendothelialgrowhfactor) и металлопротеиназы, которые взаимодействуя с рецепторами на клетках стромы, стимулируют секрецию ИЛ-6 и ФНО-α.
Характерными чертами ММ является поражение костного мозга (диффузное, диффузно-очаговое, реже очаговое), сопровождающееся костно-деструктивными изменениями (остеопороз, остеолиз) и развитие моноклональной иммуноглобулинопатии (сывороточный М-компонент и/или белок Бенс-Джонса (BJ) в моче). Синдром моноклональной иммуноглобулинопатии – парапротеинемия.
Клиническая симптоматика ММобусловлена тремя основными патогенетическими признаками заболевания:
1) инфильтрацией костного мозга клональными плазматическими клетками;
391
2)секрецией этими клетками патологического моноклонального иммуноглобулина;
3)резким снижением выработки нормальных поликлональных иммуноглобулинов.
Клинико-анатомическаяклассификация основана на данных рентгенологического исследования скелета и морфологическом анализе пунктатов и трепанатов костей, данных МРТ и КТ. Выделяют диффузно - очаговую форму (около 60% наблюдений), диффузную (24%), множественно-очаговую (15%), и редкие формы (склерозирующая (<1%) , преимущественно висцеральная (0,5%)).Стадии ММ представлены в таблице7-1.
Таблица 7-1
Стадии ММ
Стадия |
Критерии |
Опухолевая |
|
|
масса кг/м2(1012клеток~1кг |
|
|
опухолевой массы) |
I |
Совокупность следующих признаков: |
До 0,6 (низкая) |
|
Уровень Hb > 100 г/л |
|
|
Нормальный уровень Ca сыворотки |
|
|
Отсутствие остеолиза или солитарный |
|
|
костный очаг |
|
|
Низкий уровень М-компонента |
|
|
Ig J < 50 г/л |
|
|
IgA< 30 г/л |
|
|
Белок BJ в моче < 4 г/сут. |
|
II |
Показатели средние между I и II стадией |
0,6 – 1,2 (средняя) |
III |
Один или более последующих признаков: |
Более 1,2 (высокая) |
|
Уровень Hb< 85 г/л |
|
|
Уровень Ca сыворотки выше нормы |
|
|
Выраженный остеодеструктивный процесс |
|
|
Высокий уровень М-компонента: |
|
|
IgG > 70 г/л |
|
|
IgA > 50 г/л |
|
|
Белок BJ в моче > 12 г/сут. |
|
Примечание. Дополнительные стадии А и Б. А – нормальная функция почек. Б – функция почек нарушена. Hb – гемоглобин, белок BJ – белок Бенс-Джонса (Клиническая онкогематология. Под ред. М.А. Волковой, 2007)
Практически важным является определение фаз заболевания : хронической (развернутой) или острой (терминальной). Терминальн ая стадия характеризуется рефрактерностью к ранее эффективной терапии (вторичная резистентность), нарастающей миелодепрессией, прорастанием опухоли в мягкие ткани, внекостномозговыми метастазами, развитием плазмоклеточной лейкемии, иногда периферическим эритрокариоцитозом или миелией.
По степени «агрессивности» ММ (анамнез больных, динамическое наблюдение) различают:
392
-«тлеющую» ММ без признаков прогрессирования в течение многих месяцев (лет); -медленно прогрессирующую;
-быстро прогрессирующую; -«агрессивную», в т.ч. ММ-саркому и острый плазмобластный лейкоз.
Диагноз ММ основывается на 3 основных критериях (О.М. Ветякова, Е.А. Демина, 2007):
1.Плазмоклеточная инфильтрация костного мозга – плазмоцитов более 10% в стернальном пунктате.
2.Моноклональная Ig-патия (сывороточный миеломный компонент
и/или белок Бенс-Джонса в моче).
3. Выявление нарушений функций органов или систем, связанных с миеломой (одного или более):
Са (calcium) – гиперкальциемия (содержание кальция в сыворотке крови на 0,25 ммоль /л выше верхней границы нормы или более
2,75 ммоль/л),
R (renalinsufficiency) – почечная недостаточность (уровень креатинина в сыворотке крови выше 173 мкмоль/л),
А (anaemia) – анемия (содержание гемоглобина на 2 г/дл ниже нижней границы нормы или менее 10 г/дл),
В (bonelesions) – очаги лизиса в костях или остеопороз. Диагноз множественной миеломы устанавливают только при
наличии не менее 2-х из этих трех основных критериев, присутствие первого критерия обязательно.
Клиническая картина.
1. Костно-мозговой синдром. Пролиферирующие в костном мозге миеломные клетки приводят к разрушению костного вещества. В первую очередь деструктивные процессы развиваются в плоских костях и позвоночнике, иногда - в проксимальных отделах трубчатых костей (плечо, бедро); дистальные отделы конечностей и кости лицевого черепа поражаются редко.
Рентгенологические находки зависят от морфологического варианта плазмоцитомы, они убедительны при множественно-очаговой и диффузноочаговой формах и нередко отсутствуют при диффузном поражении костного мозга. Иногда миелому сопровождает развитие очагового или диффузного остеопороза.
В основе поражения костей лежит усиление резорбции костной ткани, связанные с инфильтрацией миеломных клеток, активности остеокластов и нарушением ремоделирования кости. Боли в костях отмечаются у 70% больных. Чаще всего они локализуются по ходу поражения позвонков в пояснично -крестцовой области и грудной клетке, вначале имеют мигрирующий характер, возникая только при перемене положения тела. В поздних стадиях оссалгии становятся нестерпимыми, вынуждая больных лежать неподвижно. Поражение скелета сопровождается костными деформациями. Классический симптом –
393
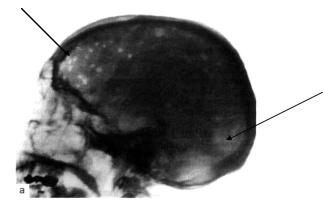
спонтанные переломы. Боль, опухоли, переломы – классическая триада Калера – типична для больных ММ в поздних стадиях и при агрессивных формах болезни. У 10% больных развивается синдром сдавления спинного мозга с нижним парапарезом и параплегией. Рентгенологически – генерализованный остеопороз, единичные или множественные очаги остеолиза, патологические переломы, иногда миелому сопровождает развитие очагового или диффузного остеосклероза.Наиболее характерны поражения в костях черепа: отдельные дефекты выглядят как бы изъеденными молью или выбитыми пробойником(рис. 7-1).
Рис. 7-1. Поражение черепа у больного ММ
Гиперкальциемия в сыворотке крови наблюдается у 10-30% больных
сММ и обусловлена усиленной резорбцией костей.
Вотличие от большинства гемобластозов, ММ поражает костный
мозг в виде дискретных очагов, между которыми сохраняются учас тки нормального кроветворения или зоны с минимальным количеством опухолевых клеток.
2. Синдром белковой патологии. Миеломная нефропатия
(парапротеинемический нефроз) – наиболее частое и серьезное проявление парапротеинемии. Поражение почек при множественной миеломе характеризуется протеинурией, постепенным снижением концентрационной функции почек. Главную роль в патогенезе поражения почек играет белок Бенс -Джонса активно реабсорбируемый из первичной мочи, повреждающий канальцевый эпителий и вызывающий постепенный склероз стромы (восходящий нефросклероз).
Амилоидоз выявляется в среднем у 1015% больных миеломной болезнью. Основным компонентом амилоидных фибрилл являются моноклональные легкие цепи или их вариабельные участки (ALамилоидоз).
В первую очередь и, главным образом, поражаются органы, богатые коллагеном: адвентиция сосудов, мышцы (сердце, язык), дерма, сухожилия и суставы. Печень, селезенка, почки обычно не страдают или амилоидные отложения в них бывают очень скромными. Атрофия роговицы при миеломной болезни могут быть следствием параамилоидных отложений в
394
соответствующих органах. В ряде случаев параамилоид образует массивные локальные псевдоопухолевые узлы по ходу желудочнокишечного тракта, иногда преимущественно откладывается в слюнных и щитовидной железах, отдельных группах лимфатических узлов.
Синдром NAMIDD – неамилоидное отложение в тканях моноклональных L- и Н-цепей или целых молекул. В отличие от амилоидоза, при NAMIDD отложения имеют аморфный нефибриллярный состав и не выявляются гистологическими методами, принятыми для диагностики амилоидоза. В их составе при помощи моноспецифических антииммуноглобулиновых антисывороток выявляются в основном L-цепи-типа. Поражаются, главным образом, почки (нефротический синдром), сердце (нарушение ритма и проводимости, снижение сократительной способности миокарда), мышцы (рабдомиолиз), ЖКТ (нарушение всасывания, диарея), кожа, суставы, слюнные железы. Описаны отложения в легких (диспноэ), верхних дыхательных путях, печени (гепатомегалия, повышение уровня трансаминаз, портальная гипертензия). Окончательный диагноз возможен только путем иммуногистологического исследования биоптатов пораженных органов.
3. Иммунодефицит синдром недостаточности антител.
Характерным симптомом множественной миеломы является резкое снижение уровня нормальных иммуноглобулинов. По мере прогрессирования заболевания их уровень закономерно снижается (гуморальный иммунодефицит закономерно нарастает) вплоть до полного их исчезновения, что выражается склонностью больных к бактериальным инфекционным осложнениям, особенно с локализацией в носоглотке, дыхательных и мочевыводящих путях.
4. Синдром повышенной вязкости характеризуется кровоточивостью слизистых оболочек, геморрагической ретинопатией, расширением вен сетчатки (fundus paraproteinaemic us), нарушениями периферического кровотока, парестезиями, синдромом Рейно, в тяжелых случаях изъязвлениями и даже гангреной дистальных отделов конечностей. Нарушения микроциркуляции в сосудах головного мозга могут служить причиной парапротеинемической комы. При криоглобулинемии эти симптомы резко выражены после охлаждения.
5. Висцеральные поражения. У 5-13% больных констатируют гепато- (или) спленомегалию. Приблизительно у половины пациентов увеличение органов связано со специфической миеломноклеточной пролиферацией, у остальных цитологический состав пунктатов и отпечатков селезенки выявляет смешанную миеломно -миелоидную или чисто миелоидную трехростковую пролиферацию. Характерным гематологическим симптомом у этих больных является миелемия (резкий левый сдвиг в лейкоцитарной формуле), нередко с эритрокариоцитозом. Поражение лимфатических узлов встречается в 0,5% случаев. Специфическое поражение желудка может проявляться в виде инфильтративного процесса, язвы или плазмоцитомы.
395
Симптоматическая периферическая нейропатия наблюдается у 5-
15% больных:нарушения тактильной и болевой чувствительности, которые сопровождаются парестезиями, мышечной слабостью, онемением конечностей, выраженным болевым синдромом. Проявления симметричны.
Гиперкальциемия встречается у 20-40% больных, чаще всего при большой массе опухоли (III стадия) и в терминальной стадии болезни, особенно при азотемии. При нормальной функции почек, достаточном потреблении жидкости гиперкальциемия не развивается. Уровень кальция резко повышается при вынужд енном обездвиживании больных. Клинически гиперкальциемия проявляется потерей аппетита, тошнотой, рвотой, нарушением стула (запором), полиурией, сонливостью, гипотонией; в далеко зашедших случаях – мышечной слабостью, потерей ориентации, судорогами, спутанностью сознания, комой. На ЭКГ регистрируются уширение комплекса QRS и зубца Т, укорочение интервала QT и ST, замедление атриовентрикулярной проводимости вплоть до блокады, иногда – фибрилляция предсердий.
Патология гемостаза. Высокая гиперпротеинемия (выше 130 г/л) часто сопровождается кровоточивостью, в патогенезе которой наряду с нарушениями тромбоцитарного и коагуляционного гемостаза, играет рольповышение вязкости крови с развитием специфического ДВСсиндрома.Тромбоцитопенические кровотечения обычно являются результатом цитостатической терапии.
Картина крови. У всех больных по мере прогрессирования болезни развивается анемия (обычно нормохромная), СОЭ увеличена не более, чем
у70% больных.
Вкартине белой крови характерных изменений нет, иногда наблюдается нейтрофилез с умеренным левым сдвигом в формуле, редко
гранулоцитопения, в отдельных случаях панцитопения. Вымывание единичных опухолевых клеток в периферическое русло – не редкость при всех формах плазмоцитомы, однако появление высокого периферического плазмоцитоза в динамике болезни, как правило, знаменует собой новую фазу опухолевой прогрессии и предвещает близкий летальный исход.
Первично лейкемические формы составляют менее 1% наблюдений. Как и при других формах парапротеинемических гемобластозов,
часто встречается абсолютный моноцитоз, реже лимфоцитоз. Эозинофилия (иногда высокая) регистрируется у 2-3% больных. На ранних стадиях болезни бывают гипертромбоцитоз и увеличение количества мегакариоцитов в пунктатах костного мозга.
Диагностический поиск.
После установления диагноза до начала лечения всем больным миеломой необходимо провести ряд обязательных исследований, которые позволяют определить форму и распространенность процесса, выявить
противопоказания |
к |
применению |
отдельных |
химиопрепаратов, |
||||
предупредить |
развитие |
патологических |
переломов, |
почечной |
||||
396
недостаточности, выявить скрыто протекающую инфекцию мочевыводящих путей и которые необходимы для последующего объективного контроля эффекта лечения.
Минимальный объем этих исследований следующий:
1)общий анализ крови с подсчетом тромбоцитов и ретикулоцитов;
2)общий анализ мочи;
3)определение концентрационной способности почек по методу
Зимницкого;
4)при наличии протеинурии – определение суточных потерь белка с
мочой;
5)исследование уровней креатинина, мочевой кислоты, кальция сыворотки крови;
6)определение содержания в сыворотке билирубина, холестерина,
трансаминаз.
7)определение общего белка сыворотки крови;
8)электрофорез сывороточных белков и определение содержания
белка в М-компоненте;
9)электрофорез белков концентрированной мочи;
10)рентгенография всех костей скелета, кроме дистальных отделов конечностей (от середины плеча до кисти и от середины бедра до стопы, если нет клинических признаков поражения костей в этих областях);
11)стернальная пункция – основной метод верификации диагноза.
К факторам высокого риска относятся следующие клинические и лабораторные показатели перед началом лечения:
1)выраженная миелодепрессия;
2)быстрый (в течение недель) рост опухоли, определяемый при динамическом рентгенологическом исследовании костных деструкций или
(реже) визуально и пальпаторно;
3)стремительное нарастание уровней сывороточного и/или мочевого парапротеина;
4)наличие мягкотканных метастазов;
5)плазмоклеточная лейкемия;
6)миелемия (резкий сдвиг в лейкоцитарной формуле, эритрокариоцитоз);
7)высокий индекс метки опухолевых клеток (L1> 1,2%);
8)процент плазматических клеток в S-фазе > 3 (методом проточной цитометрии);
9)β2микроглобулин >верхней границы нормы;
10)высокий уровень ИЛ-6 и рецепторов к ИЛ-6;
11)низкий уровень ИЛ-2;
12) ЛДГ >300 ед/л;
13)цитогенетические аномалии: гиподиплоидия, делеция или утрата 13 хромосомы и др.;
14)иммунофенотип CD20+++, СД 56(-), CD28++;
15)СРБ > 6 мг/л;
397
16) альбумин 30 г/л и менее.
Дифференциальный диагноз.
Макроглобулинемия Вальденстрема - редко встречающееся состояние, характеризующееся полным набором признаков гаммапатии, содержанием IgM моноклонального протеина, но без поражения костей.
Болезни тяжелых цепей - опухолевые В-лимфопролиферативные заболевания, характерной особенностью которых является секреция фрагментов Н-цепей различных классов Ig. Диагностика основывается на
иммунохимическом анализе сывороточных белков. |
|
|
||
Моноклональные иммуноглобулинопатииДиагностируется |
у |
|||
носителей |
моноклональной |
иммуноглобулинопатии |
методами |
|
электрофореза и иммунохимического анализа Ig после исключения ММ. Солитарные плазмоцитомы (костные и внекостные) – чаще всего
встречаются в носоглотке и в верхних дыхательных путях, а также по ходу желудочно-кишечного тракта. Чаще всего встечаются у мужчин, средний возраст на 10 лет меньше, чем при ММ. Диагноз солитарных плазмоклеточных опухолей должен быть доказан морфологически (биопсия, пункция); объем исследований, необходимых для выявления возможной генерализации такой же, как и при постановке диагноза ММ.
Исключая истинно реактивные, преходящие формы парапротеинемии, все пациенты с моноклональнойIg-патией должны считаться угрожаемыми по парапротеинемическому гемобластозу в течение многих лет.
Цитостатическая противоопухолевая терапия.
Показания: признаки нарастания опухолевой массы (падение показателей красной крови, повышение сывороточного и/или мочевого парапротеина, развитие болевого синдрома).
Выбор метода терапии.
1.Стационарная химиотерапия.
2.Локальное облучение очагов миеломы.
3.Высокодозная химиотерапия с трансплантацией костного мозга или стволовых клеток периферической крови.
Программы химиотерапии.
А. Программная терапия умеренными дозами с поддерживающим лечением ударными прерывистыми дозами.
Б. Ударная прерывистая терапия: МР,СР. В. Полихимиотерапия.
МР – алкеран + преднизолон. Перерыв 4 недели, далее поддерживающая терапия.
СР – циклофосфан + преднизолон. Перерыв 3-4 недели, далее поддерживающая терапия.
Протокол М2: винкристин , алкеран (мелфалан) , циклофосфан, кармустин (или бемостил) и преднизолон.
М2 + адриабластин.
398
VAD – винкристин, адриабластин, дексаметазон. VAMP – винкристин, адриабластин, метилпреднизолон.
Преодоление резистентности к полихимиотерапии.
Пульс – терапия дексаметазоном.
VID – винкристин, идарубицин, дексаметазон.
Циклоплатам(производное платины III поколения) – медиана продолжительности эффекта 6 месяцев.
Интенсивная терапия ММ (High-dosechemotherapy – HDC) – раннее использование высокодозной химиотерапии мелфаланом с аутотрансплантацией стволовых клеток периферической крови или костного мозга.
Современная тенденция в терапии ММ –использование преставителя группы ингибиторов протеосом – бортезомиба. Бортезомиб обладает высокой степенью сродства к протеолитической оболочке, замедляющей действие протеосом. Ингибирование протеосомы приводит к снижению уровня определенных регуляторных белков, которые поддерживают гомеостаз неопластов, тем самым, приводя к гибели опухолевой клетки.
Препарат второй линии терапии при резистентности и/или рецидивирующем течении ММ – леналидомид.
Новые стратегии в лечении ММ.
PAD – бортезомиб + доксорубицин + дексаметазон.
PCD–бортезомиб + циклофосфамид + дексаметазон.
Критерии эффективности цитостатической терапии ММ.
Объективное улучшение регистрируется при наличии одного из следующих показателей, сохраняющихся в течение 2 месяцев и более:
1.Снижение концентрации парапротеина в сыворотке более чем на 50% (до уровня < 40 г/л);
2.Снижение экскреции белка Бенс-Джонса, более чем на 50% (< 0,5
г/сут) от исходного уровня;
3.Уменьшение площади опухолей, определяемой произведением двух наибольших диаметров, на 50% и более;
4.Появление рентгенологических признаков заживления костных деструкций.
Лечение считается эффективным только у тех больных, которые
имеют стабильные или увеличивающиеся показатели красной крови (Hb>90 г/л), сывороточного альбумина (> 30 г/л) и, у которых количество и размеры остеодеструктивных очагов не нарастают, а уровень кальция не превышает нормы.
Лечение осложнений ММ.
Локальная лучевая терапия показана во всех случаях угрозы патологических переломов в опорных частях скелета (позвоночник, крестцово-подвздошная область, лонные, седалищные кости, тело подвздошной кости над вертлужной впадиной, бедренные мало -, большеберцовые и плечевые кости), даже при отсутствии болевого
399
синдрома. Локальное облучение используется при наличии органических опухолевых узлов в костях и мягких тканях, радикулярных болях, связанных со сдавливанием корешков спинного мозга опухолью или компрессированными телами позвонков, на начальных этапах сдавливания спинного мозга.
Лечение и профилактика осложнений.
Антибактериальная терапия инфекционных осложнений проводится по общим правилам. Лечение больных с иммунодефицитом. Преимущество отдается цефалоспоринам III и IV поколения (фортум, максипим), карбапенемам (тиенам, меронем), "защищенным" пенициллинам широкого спектра (тазоцин); при необходимости применя - ют ванкомицин и аминогликозид с минимальной нефротоксичностью - нетромицин. Антибактериальная терапия сочетается с антигрибко выми препаратами (низорал, дифлюкан).
Следует избегать применения при ММ антибактериальных средств, обладающих нефротоксично стью (гентамицин, стрептомицин, канамицин и др.).
Лечение почечной недостаточности, обусловленной миеломой – адекватная химиотерапия (VAD) и гидратация. Больным с тяжелой уремией показан гемодиализ. При высоком уровне моноклонального белка
– плазмаферез.
При глубокой анемии необходимы трансфузии эритроцитарной массы, рекомбинантный эритропоэтин для улучшения качества жизни
больных. |
|
|
|
Основными |
методами |
ликвидации |
|
гиперкальциемииявляютсяхимиотерапия |
и |
глюкокортикостероиды. |
|
Дополнительную роль играет гипергидратация. |
|
||
Для профилактики развития и лечения костных осложнений у больных ММ используются бисфосфонаты: памидронат динатрия (аредия), золедронат (зомета).
Лечение патологических переломов при миеломе не должно отличаться от лечения переломов костей у здоровых людей.
Всем больным, в том числе находящимся на постельном режиме, постоянно проводят лечебную физкультуру, обеспечивающую нагрузку на опорные части скелета. Постельный режим нежелателен, его назначают только при острых болях, в связи со свежими переломами костей.
Ношение корсетов не рекомендуется, за исключением послеоперационного периода у больных с декомпрессивной ламинэктомией.
При синдроме высокой вязкости, кровоточивости, при гиперпротеинемии 130-140 г/л показан плазмаферез.
Высокий риск синдрома распада опухоли – аллопуринол 300-600 мг/сутки в первые 5 – 7 дней каждого курса.
Прогноз. Длительность жизни в большой степени зависит от чувствительности опухоли к цитостатической терапии.
400