

В результате переноса бензоил-СоА на глицин происходит образование коэнзима А и гиппуровой кислоты, которая выводится с мочой.
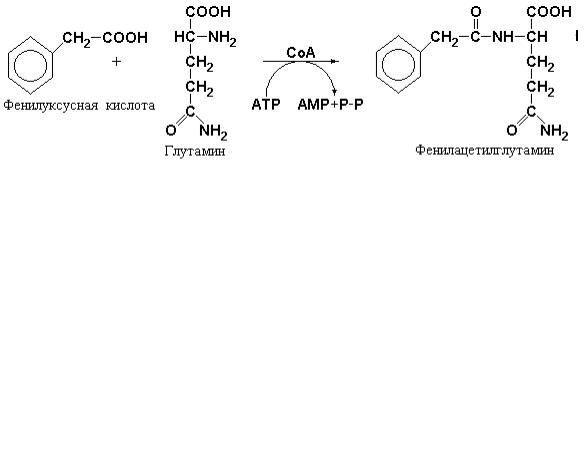
Сходная реакция имеет место между гомологом бензойной кислоты – фенилуксусной кислотой и глутамином. Отличие этой реакции заключается в отсутствии накопления интермедиата, представляющего собой тиоэфир с CoA. Тем не менее реакция также зависит от присутствия CoA, но карбоксильная группа фенилуксусной кислоты взаимодействует с α- аминогруппой глутамина.
Эти реакции не имеют большого значения при нормальном метаболизме, но могут играть большую роль при введении ароматических кислот с целью удаления избытка NH4+ при нарушениях орнитинового цикла
(Brusilow S.W., Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis, New Engl. J. Med., 1984, 310, 1630-1642).
Наследственные дефекты ферментов цикла мочевины
Значение нормального функционирования цикла мочевины в организме человека трудно переоценить. Известно, что новорожденные с полным отсутствием одного или нескольких ферментов цикла живут всего в течение нескольких дней. Многие синдромы, связанные с дефицитом тех или иных ферментов были идентифицированы и описаны благодаря тому, что эти ферменты все же проявляли некоторую активность. Проведение аналогии с другими мутациями позволяет сделать вывод, что в первую очередь нарушения активности ферментов цикла мочевины связаны с изменением величины Km, но не Vmax. Однако охарактеризовать более подробно мутации генов, кодирующих эти важнейшие белки у человека
довольно сложно (Grisolia S., Baguena R., Mayor F., The urea cycle, New York: Wiley, 1976).
Вместе с тем, в настоящее время уже охарактеризованы основные синдромы, связанные с дефицитом каждого из ферментов цикла мочевины. Разрыв цикла в какой-либо точке по-разному влияет на метаболизм азота, поскольку некоторые интермедиаты могут диффундировать из гепатоцитов, накапливаться в крови и попадать в мочу. Поэтому симптомы, прогнозы и терапия при дефиците различных ферментов отличаются. В целом описано несколько довольно часто встречающихся заболеваний, которые проявляются в задержке умственного развития или сопровождаются комой и смертью в раннем возрасте.
N-ацетилглутамат синтетаза
Значение активатора карбамоилфосфат-синтетазы было обнаружено при обследовании новорожденных с гипераммониемией и общей гипераминоацидемией. У этих новорожденных клетки печени не обеспечивали какого-либо заметного синтеза N-ацетилглутамата. В таких случаях нормальный обмен азота можно поддерживать с помощью диеты, содержащей низкое количество белка и введением карбамоилглутамата – аналога N-ацетилглутамата, который также служит активатором
карбамоилфосфат синтетазы.
Карбамоилфосфат синтетаза
В тех случаях, когда у младенцев уровень синтеза карбамоилфосфат синтетазы составляет 0-50% от нормального содержания фермента в гепатоцитах также отмечается развитие гипераммониемии. Этим младенцам вводят бензойную кислоту с фенилацетатом, которые достаточео эффективно снимают проявления гипераммониемии (Brusilow S.W.,
Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis, New Engl. J. Med., 1984, 310, 1630-1642). Весьма полезным является добавление в низкобелковую диету аргинина, который стимулирует синтез N-ацетилглутамата, а последний в свою очередь обеспечивает образование достаточного количества карбамоилфосфата даже при низком уровне фермента в клетках печени. Вообще, нехватка карбамоилфосфат синтетазы сопровождается задержкой умственного развития, которое может быть следствием гипераммониемии в течение определенного контролируемого периода.
Орнитин транскарбамоилаза
Большая часть заболеваний, связанных с нарушениями цикла мочевины определяется дефицитом или утратой орнитин
транскарбамоилазы. Ранняя смерть может быть предотвращена удалением избытка аммиака, а дальнейшее его накопление устраняется теми же мероприятиями, которые предпринимаются при лечении заболевания, связанного с нехваткой карбамоилфосфат синтетазы.
Генетический анализ повреждения орнитин транскарбамоилазы показал, что ген этого белка расположен в Х-хромосоме. Поэтому данное заболевание чеще встречается у мальчиков, чем у девочек, у которых в гетерозиготном состоянии повреждение гена не приводит к проявлению симптомов болезни. Кроме аммиака и аминокислот в крови повышается содержание оротовой кислоты. Появление оротата объясняется тем, что избыток карбамоилфосфата проникает в цитоплазму гепатоцитов, где он взаимодействует с аспарагиновой кислотой и приводит к накоплению оротовой кислоты.
Аргининосукцинат синтетаза
Нехватка данного фермента цикла мочевины приводит к накоплению цитруллина в крови, что сопровождается цитруллинемией. В некоторых случаях большая часть азота выводится именно в виде цитруллина. В дополнение к ограничению содержания белка в диете, терапия этого заболевания основана на дополнительном введении аргинина, необходимого для синтеза белка и синтеза креатина и орнитина, потребность в котором определяется не только функционированием цикла мочевины.
Аргининосукцинат лиаза
При этом виде нарушений работы орнитинового цикла у пациентов с мочой выводится очень большое количество аргининосукцината. Данное заболевание протекает с таким количеством симптомов, что часто бывает сложно подобрать эффективное лечение. Тем не менее, ограничение белка, введение бензойной кислоты, фенилацетата и аргинина довольно часто приводит к желаемому результату (Brusilow S.W., Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis, New Engl. J. Med., 1984, 310, 1630-1642).
Аргиназа
Дефицит аргиназы представляет собой очень редкое заболевание, которое сопровождается различными и многочисленными нарушениями развития и функций центральной нервной системы. При этом заболевании в больших количествах накапливается и выводится не только аргинин, но и его предшественники, а также продукты обмена.
Терапию проводят бензоатом натрия и кетоаналогами незаменимых аминокислот (Brusilow S.W., Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis, New Engl. J. Med., 1984, 310, 1630-1642).