

25. Химия металлов (Ме) и металлических плёнок
Осаждение гальванических покрытий, в основе которого лежит реакция катодного восстановления, в основном применяется для создания омических контактов и нанесения электродов. Создание контакта Ме-п/п (невыпрямляющий контакт) осуществляется методом химического никелирования - каталитического восстановления Niиз раствора его соли на пластине п/п (среда щелочная рН = 7÷8), при этом происходят следующие реакции: 1)Ni2++Me→Ni++Me+(электрон от Ме-катализатора перешел наNi); 2) 2Ni→Ni+Ni2+(диспропорционирование); 3)OH-+Me+→OH+ +Me(возвращение электрона к Ме-катализатору ионом гидроксила); 4)H2PO2- + + 2OH→H2PO3-+H2O(радикал ОН восстанавливается гипофосфитом). Суммарная реакция:Ni2++H2PO2-+ 2OH-→Ni+H2PO3-+ +H2O.
Улучшение адгезии Niи уменьшение сопротивления контакта после осажденияNiведётся термообработкой в вакууме 5∙10-8÷1∙10-7Па в течение 15÷20 минут при 500÷700оС дляSi, скорость охлаждения (во избежания механических напряжений) - 10град/мин. Затем проводится вторичное никелирование, затем нанесение золотого покрытия для присоединения электрического вывода методом пайки или термокомпрессии. ПреимуществаAu-покрытия: кислотостойкость, большая электро- и теплопроводность, простота пайки.Au-покрытие получается гальваническим золочением в цианистых или железистосинеродистых электролитах.
Создание балочных электродных выводов на диодных структурах на исходной Siпластине с защитным слоемSiO2включает следующие операции: 1) созданиеp+-анодных иn+-катодных областей; 2) с помощью фотолитографии вытравливание в слоеSiO2отверстий для для создания омических контактов; 3) методом термического напыления создание омических контактов; 4) напыление двухслойного Ме-покрытия (слоя, осуществляющего сцепление, и токопроводящего слоя для гальванического покрытия); 5) защита маской фоторезиста; 6) осаждениеAu-Выводов (гальваническим путём); 7) фоторезист и биметаллическое покрытие удаляют катодным распылением.
Создание омических контактов (ОК) соединений AIIIBV- наиболее распространены ОК на основеAu, создаваемые методом термического распыления в вакууме: к слоямn-типа проводимости применяются сплавыAu+ 5÷25%Ge,Au+ 4÷10%Si; к слоямp-типа проводимости – сплавAu+ + 1÷25%Zn. СплавыAu-GeиAu-Si– основные для соединенийGaAs,GaAsP,GaP, иGaInP, напыление проводится при 200-300оС, затем проводят вжигание при оптимальной для соединения температуре. Поверх сплава наносятNi(иногдаNiвводят в сплав – до 5%). Омический контакт проводят после отжига:Ga0,6Al0,4As- при 350оС,GaAl0,6P0,4- 400oC,GaP– - 500oC. СплавыAu-Zn– хороший контакт без вжигания только дляGaP. ДляGaAlиInPсn-типа проводимостью применяются сплавыAg-Ge-InиAg-Ge-Ni(8:1:1), вжигание при 580оС (1 минута). ДляGaAsPиGaAlAsс р-типа проводимостью применяется контакт изAl(1÷2мкм), для второго соединения применяется отжиг при 500оС. Реже для последних соединений применяются сплавыAg-Sn(1:2, 1:4), вжигание при 440÷450оС, 8÷10 минут.
Лабораторная работа №1
ПРИГОТОВЛЕНИЕ ЛАКА, ЛЮМИНОФОРНОЙ СУСПЕНЗИИ, НАНЕСЕНИЕ ПОКРЫТИЙ.
(Продолжительность лабораторной работы 6 ч; домашняя подготовка 4 ч) Приборы и принадлежности: установка для сушки люминофорного слоя, установка для проверки светопропускания, валковая мельница, форфоровый барабан с шарами, вискозиметр ВЗ-4, набор ареометров. Материалы: люминофор, смола БМК-5, бутилацетат, ацетон, капроновое сито.
ЗАДАНИЯ К РАБОТЕ.
1. Ознакомиться с теоретическим материалом. 2. Приготовить лак из смолы БМК-5 и люминофорную суспензию. 3. Измерить удельный вес суспензии, довести удельный вес до 1,32÷1,52 г/см3 бутилацетатом. 4. Сформировать люминофорное покрытие на трубках-колбах. 5. Измерить толщину люминофорного слоя. 6. Измерить светопропускание слоя. 7. Произвести термическую обработку люминофорного слоя. 8. Измерить удельную нагрузку люминофора на трубках. 9. Определить плотность упаковки люминофорных частиц в слое. УКАЗАНИЯ К ВЫПОЛНЕНИЮ РАБОТЫ
По п.2.Основными компонентами люминофорного покрытия на внутренней поверхности трубок-колб являются люминофор, связующее вещество и растворитель. В качестве связующего вещества используется смола БМК-5. Густой лак готовится растворением смолы БМК-5 в бутилацетате (на 500г смолы — Зл бутилацетата). Густой лак разводится бутилацетатом до рабочей вязкости 10спз (0,01Пас). В фарфоровый барабан емкостью 2л с 600г ситалловых шаров диаметром 18÷20мм загружаются следующие компоненты: 100г люминофора, 60мл лака (0,01Пас) и тетрафосфат бария – 1÷1,5г. ТФБ можно добавить в виде суспензии. С этой целью 250г тетрафосфата бария вместе с лаком БМК-5 вязкостью 10спз с добавлением 0,04г лимонной кислоты размалывают в фарфоровом барабане с 600г шаров в течение 15 час.Размол смеси люминофора, лака и ТФБ проводят 15÷20 мин. Суспензию сливают в стакан, профильтровывают через капроное сито и измеряют уд. вес. При необходимости уд. вес. доводится бутилацетатом до 1,32÷1,52 г/см3.По п.4.Для формирования люминофорного покрытия на внутренней поверхности трубок применяется метод полива люминофорной суспензиией.По п 5. Микроскопом измерить толщину люминофорного слоя.По п.6.На установке измерить величину светопропускания люминофорного слоя (в отн. ед).По п.7.Произвести термообработку люминофорного покрытия трубки при Т = 450÷500оС в течение 1÷2 мин.По п.8.Для определения удельной нагрузки на трубках люминофор аккуратно счищают с колбы, взвешивают и делят на величину поверхности. Она должна находиться в пределах 3÷5 мг/см2.По п.9.Плотность упаковки частиц в люминофорном покрытии определяется по формуле:y=d/(nh) (1.1) гдеп - удельная нагрузка люминофорного покрытия, г/см2;h -толщина люминофорного слоя, см;d - удельный вес люминофора ГФК: 3,25 г/см3. Плотность упаковки частиц колеблется от 32 до 44%, что объясняется различными поверхностными свойствами люминофора. Кроме того, плотность упаковки частиц люминофора в покрытии определяется и различными адгезионными взаимодействиями люминофора и сополимера. Установлено, что с увеличением плотности упаковки улучшается стабильность светового потока люминесцентных ламп. Например, повышение плотности на 6% увеличивает световой поток ламп на 2÷3%.Контрольные вопросы 1.Какие соединения могут быть основой люминофоров различного применения?
2. Какие элементы являются активаторами в люминофорах и каковы спектры излучения последних?
3. Каковы функции биндеров (связующих) в люминофорных суспензиях и какие полимеры используются в них?
4. Почему удельный вес суспезии и удельная нагрузка люминофора должны быть оптимальными и как их определить?
5. Каковы технологии изготовления лака и суспензии с различными связующими?
6. Зачем, когда и по каким режимам проводятся термические обработки люминофорного слоя.
7. Как определяется удельная нагрузка люминофора на трубках.
8. Роль гранулометрии люминофора в формировании равномерного по толщине слоя.
Лабораторная работа 2
ПРИГОТОВЛЕНИЕ ЛАКА ФТОРПОЛИМЕРА И НАНЕСЕНИЕ ЗАЩИТНОГО ПОКРЫТИЯ НА ПОВЕРХНОСТЬ ЛАМП.
(Продолжительность лабораторной работы 4 ч; домашняя подготовка 4 ч) Приготовление лакового раствора ФЛ-32Л осуществляется в две стадии. На первой стадии проводится растворение полимера в изоамилацетате с образованием густой однородной массы. Затем для приготовления раствора полимера рабочей вязкости при комнатной температуре проводилось разбавление густого лака бутилацетатом.
Существенное влияние на бездефектное формирование защитных покрытий и обеспечение достаточной адгезии к стеклу и цоколю оказывает чистота поверхности ламп. Поэтому перед нанесением защитного покрытия необходимо очистить и обезжирить органическими растворителями покрываемые поверхности.
По результатам исследований был разработан следующий технологический режим нанесения защитных, ударопрочных покрытий: метод нанесения - окунание; вязкость по ВЗ-4 при 18÷20оС — 16÷30мин; число наносимых слоев - 1 и более; общая толщина покрытия, мм - 0,05÷ ÷0,50; температура сушки покрытия,оС - 60÷90; время сушки слоя, мин - 15÷25. Достаточная механическая прочность достигается при толщине пленки от 0,05мм. Коэффициент пропускания такой пленки составляет 0,98. При падении на твердую поверхность с высоты 1÷2м разрушения стекла колбы не вызывает разрыва пленки.
ЗАДАНИЯ К РАБОТЕ
1. Приготовить лак на основе полимера Ф-32Л.
2. Нанести защитное покрытие на колбу и цоколь лампы. 3. Произвести сушку покрытия. 4. Измерить толщину покрытия. УКАЗАНИЯ К ВЫПОЛНЕНИЮ ЗАДАНИЯ. По п.1.В фарфоровый барабан емкостью 2л засыпать 200г полимера и добавить 400мл изоамилацетата. Раствор перемешать на валковой мельнице в течение 3÷4ч, затем добавить 300мл бутилацетата и перемешать еще 10÷15мин. Слить раствор в стакан, измерить условную вязкость. Бутилацетатом довести условную вязкость до рабочей вискозиметром ВЗ-4.По п.2.Методом окунания в емкость с раствором полимера сформировать покрытие на колбе лампы.
По п.3.Поместить изделие с покрытием в держатель сушильного шкафа с температурой 60÷80оС на 20÷30мин.
По п.4.Микроскопом замерить толщину покрытия.
Контрольные вопросы
1. Каковы основные свойства лаковых фторполимеров и особенности технологии получения лака разной вязкости из порошка полимера?
2. Каким образом можно получить защитные покрытия разной толщины из фторполимера Ф-32Л?
3. Назовите режимы сушки покрытия из фторполимера.
4. Как измерить толщину слоя фторполимера.
5. Назначение пленочных защитных покрытий.
6. Химические свойства фторполимероф для защитных покрытий.
7. Почему нельзя нагревать фторполимеры выше 250оС?
Лабораторная работы №З
АНАЛИЗ ПЛАТИНИТА [3]
(Продолжительность
лабораторной работы 4 ч; домашняя
подготовка 4 ч)
ЗАДАНИЯ К РАБОТЕ:
1.
Определение бората.
2. Определение
меди.
3. Определение никеля.
4. Анализ
дефектов.1. Определение бората.
Сущность метода.
Находящийся
на поверхности платинита борат снимают
кипячением образца в 1% растворе винной
или лимонной кислоты и по потере веса
образца определяют процентное содержание
бората.Необходимые реактивы и
растворы: 1.Кислота винная или
лимонная, ч.д.а., 1%-ный раствор.
2. Спирт
этиловый, ректифицированный.Ход
анализа Измерить средний диаметр
проволоки, взятой для анализа. Отрезок
платинитовой проволоки весом 0,5г
сматывают в моток (свободная намотка),
опускают в стакан, содержащий 50-60мл
нагретого 1% раствора лимонной кислоты
и кипятят в течение 10 мин. Затем проволоку
вынимают, промывают вначале в воде,
потом в спирте и сушат в сушильном шкафу
при температуре 70÷80оС до постоянного
веса. Поверхность слоя меди должна быть
светлой и чистой. Полноту снятия бората
проверяют повторным кипячением проволоки
в растворе лимонной кислоты в течение
5 мин. Приполном снятии бората вес
проволоки после повторного кипячения
не изменяется. Процентное содержание
бората калия по формуле:
Х = ((а-в)/а) ·100% ,
(3.1)
где а -
навеска пробы платинита, г; в -
вес пробы платинита после снятия
бората, г.2. Определение меди.
Сущность метода.
Перед началом
анализа определить средний размер
диаметра проволоки. После снятия буры
проволоку растворяют в концентрированном
аммиачном растворе хлорной меди с
добавлением хлористого аммония. Медь
переходит в раствор вследствие окисления
ее ионамиCu с
образованием растворимой комплексной
соли. Содержание меди определяют по
разности весов проволоки до и после
обработки ее в растворе хлорной меди. Необходимые реактивы
и растворы: 1. Аммиак водный, ч.д.а,
25%-ный.
2. Спирт этиловый.
3. Медь
хлорная, аммиачный раствор: 30г хлорной
меди, ч.д.а., растворяют при непрерывном
перемешивании в 70мл 25%-ного аммиака с
добавлением 20г хлористого аммония,
ч.д.а.Ход анализа Проволоку
после снятия буры взвешивают, помещают
в стакан емкостью 100мл, наливают раствор
хлорной меди в таком количестве, чтобы
вся проволока была покрыта раствором,
и оставляют стоять до полного растворения
меди (30÷40 мин). Весь слой меди с проволоки
переходит в раствор. Оставшаяся проволока
представляет собой сплав железа и
никеля. Ее промывают в теплой воде с
небольшим количеством аммиака и затем
в спирте, сушат и взвешивают. Разница
в весе соответствует весу меди. Процентное
содержание меди вычисляют по формуле:
М =
(А/а)100%,
(3.2)
где М - определяемый
компонент;А-
вес осадка, г, а -
навеска пробы, г. 3. Определение
никеля. Сущность метода.
Никель
определяют весовым методом, основанным
на осаждении его диметилглиоксимом в
слабоаммиачной среде. Железо удерживается
в растворе лимонной кислотой в виде
растворимых комплексных соединений.
С никелем диметилглиоксим в аммиачной
среде образует розово-красный осадок
состава.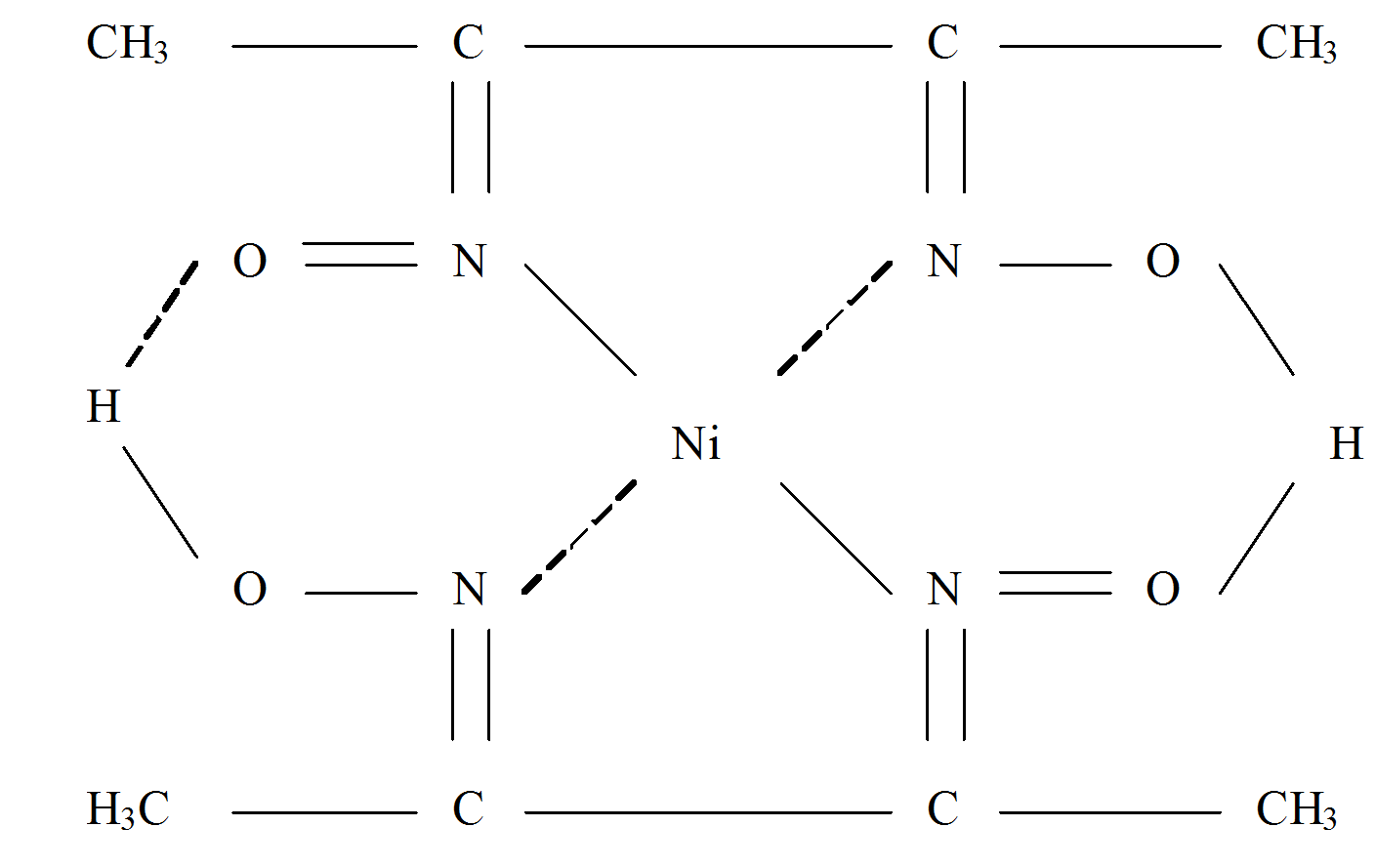 Необходимые
реактивы и растворы: 1. Кислота
азотная, ч.д.а., пл.1,40, разбавленная 1:3.
2. Кислота винная или лимонная ч.д.а.,
10%-ый раствор.
3. Аммиак водный, ч.д.а.,
25%-ый и разбавленный 1:1.
4. Аммоний
надсернокислый, ч.д.а., 1%-ый раствор.5.
диметилглиоксим, ч.д.а., 1%-ый спиртовой
раствор.
6. Соляная кислота, ч.д.а.,
пл.1,19, разбавленная 1:1.Ход анализа
Перед началом анализа определить
средний размер диаметра проволоки.
Навеску 0,1г проволоки, освобожденной
от буры и меди, растворяют при нагревании
в 20÷30мл азотной кислоты, разбавленной
1:1 в стакане емкостью 400-500мл, затем
кипятят до прекращения выделения
окислов азота. После растворения пробы
прибавляют в раствор 25мл 25%-ного раствора
лимонной кислоты для удержания в
растворе железа, нейтрализуют раствор
аммиаком по бумажке Конго, при постоянном
перемешивании прибавляют несколько
капель соляной кислоты (1:1) до кислой
реакции. Раствор разбавляют горячей
водой до объема 250÷300мл и прибавляют 70
мл диметилглиоксима, после чего приливают
аммиак до слабого запаха (1÷2мл). Осадку
дают отстояться на теплом месте 20÷30
мин, фильтруют через фильтр с красной
лентой, промывают на фильтре теплой
водой до полного обесцвечивания
промывных вод. Промытый влажный осадок
глиоксимата никеля вместе с фильтром
помещают во взвешенный фарфоровый
тигель. Осторожно высушивают, не допуская
воспламенения. Затем осадок прокаливают
в муфеле при 750÷800оС до постоянного
веса.
Необходимые
реактивы и растворы: 1. Кислота
азотная, ч.д.а., пл.1,40, разбавленная 1:3.
2. Кислота винная или лимонная ч.д.а.,
10%-ый раствор.
3. Аммиак водный, ч.д.а.,
25%-ый и разбавленный 1:1.
4. Аммоний
надсернокислый, ч.д.а., 1%-ый раствор.5.
диметилглиоксим, ч.д.а., 1%-ый спиртовой
раствор.
6. Соляная кислота, ч.д.а.,
пл.1,19, разбавленная 1:1.Ход анализа
Перед началом анализа определить
средний размер диаметра проволоки.
Навеску 0,1г проволоки, освобожденной
от буры и меди, растворяют при нагревании
в 20÷30мл азотной кислоты, разбавленной
1:1 в стакане емкостью 400-500мл, затем
кипятят до прекращения выделения
окислов азота. После растворения пробы
прибавляют в раствор 25мл 25%-ного раствора
лимонной кислоты для удержания в
растворе железа, нейтрализуют раствор
аммиаком по бумажке Конго, при постоянном
перемешивании прибавляют несколько
капель соляной кислоты (1:1) до кислой
реакции. Раствор разбавляют горячей
водой до объема 250÷300мл и прибавляют 70
мл диметилглиоксима, после чего приливают
аммиак до слабого запаха (1÷2мл). Осадку
дают отстояться на теплом месте 20÷30
мин, фильтруют через фильтр с красной
лентой, промывают на фильтре теплой
водой до полного обесцвечивания
промывных вод. Промытый влажный осадок
глиоксимата никеля вместе с фильтром
помещают во взвешенный фарфоровый
тигель. Осторожно высушивают, не допуская
воспламенения. Затем осадок прокаливают
в муфеле при 750÷800оС до постоянного
веса.
Процентное содержание никеля вычисляют по формуле: М= ((А· F)/а) 100%, (3.3) где М - определяемый компонент, А - вес осадка, г, а — навеска пробы, взятая для определения, г,F- пересчетный фактор. Фактор пересчетаFNiO наNi равен 0,7858.
Определить условный процент содержания меди по формуле, приведенной выше. Контрольные вопросы1. Где применяется платинитовая проволока и каковы её физико-термические свойства.
2. Каковы функции слоя борнокислого калия (бората) на слое из меди.
3. Каковы методы определения бората?
4. В чем заключается сущность меди в платинитового содержания меди в платинитовой проволоке?
5. В чем заключается сущность определения весового содержания никеля в платенитовой проволоке.
6. Какие дефекты платинитовой проволоке Вы знаете?
Лабораторная работы № 4 АНАЛИЗ ХИМИЧЕСКОЙ УСТОЙЧИВОСТИ СТЕКОЛ (Продолжительность лабораторной работы 6 ч; домашняя подготовка 4 ч) ЗАДАНИЕ К РАБОТЕ 1. Определение гидролитической устойчивости стекла. 2. Определение устойчивости стекла по отношению к кислотам. 3. Определение устойчивости стекла по отношению к щелочам. УКАЗАНИЯ К РАБОТЕ 1.Определение гидролитической устойчивости стекла Сущность метода заключается в том, что навеску порошка исследуемого стекла кипятят в течение определенного времени в дистиллированной воде. Содержание щелочей определяют титрованием 0,1н HCl. По количеству затраченной кислоты определяют содержание Na2O в воде. По полученным результатам характеризуют класс стекла. Ход анализа Пробу стекла для анализа промывают в воде и высушивают в сушильном шкафу при температуре 105÷110оС. Затем стекло дробят, растирают в агатовой ступке до размеров зерен 0,3-0,5мм, помещают в стакан с притертой пробкой и сушат в течение часа при 100÷110оС и из него берут навеску для анализа. 2г порошка помещают в стакан из особо устойчивого стекла и кипятят в течение 1 часа в 100мл дистиллированной воды. Отделяют раствор от осадка и титруют 0,1н HCl. При данных условиях опыта 1см3 0,1н HCl соответствует 0,31мг Na2О. Класс стекла определить по табл.Л4-1.
2. Определение устойчивости стекла
по отношению к кислотам. Сущность
метода.
Сущность метода
заключается в нахождении потери веса
образца после обработки его в растворе
кислот.Ход анализа Поскольку
стекло менее подвержено действию
кислот, чем щелочей поверхность образца
должна быть больше, чем при испытании
в щелочных растворах. Общая поверхность
пластинок или трубок должна быть около
200см3. После промывания образца
в дистиллированной воде и спирте,
образцы сушат и взвешивают с точностью
до 0,1мг. Затем кипятят в течение 2 час в
соляной кислоте (20 вес %, пл.1,1г/см3).
После повторного промывания и сушки,
определяют потерю веса образцов,
отнесенную к 100см2поверхности.
Класс устойчивости стекла по отношению
к кислотам определить по табл.Л4-2.
Гидролитические классы стёкол
Таблица Л4-1
Классы
устойчивости стёкол по отношению к
кислотам Таблица Л4-2

3. Определение устойчивости стекла по отношению к щелочам Сущность метода заключается в потере веса образца после обработки его поверхности раствором щелочи. Ход анализа Образец стекла в виде пластины или трубки, поверхность которого составляет 10÷20см2 и определена с точностью до 5%, после промывки в дистиллированной воде и спирте тщательно просушивают и взвешивают с точностью до 0,1мг. Затем образец в течение 2 час выдерживают в кипяшем растворе едкого натра (20,5г) и карбоната натрия (27г) в 1 л дистиллированной воды. Углекислый газ сильно влияет на ход реакции щелочи со стеклом. Кроме того, его присутствие приближает условия испытания к реальным. После обработки образец нейтрализуют кратковременным погружением в сильно разбавленный раствор соляной кислоты, промывают дистиллированной водой, сушат и снова взвешивают. Зарегистрированная разница в весе, отнесенная к 100см2 поверхности служит основой для классификации стекол по степени устойчивости в растворах щелочей. Класс устойчивости стекол по отношению к щелочам см. в табл.Л4-3.
Классы
устойчивости стёкол по отношению к
щелочам Таблица Л4-3
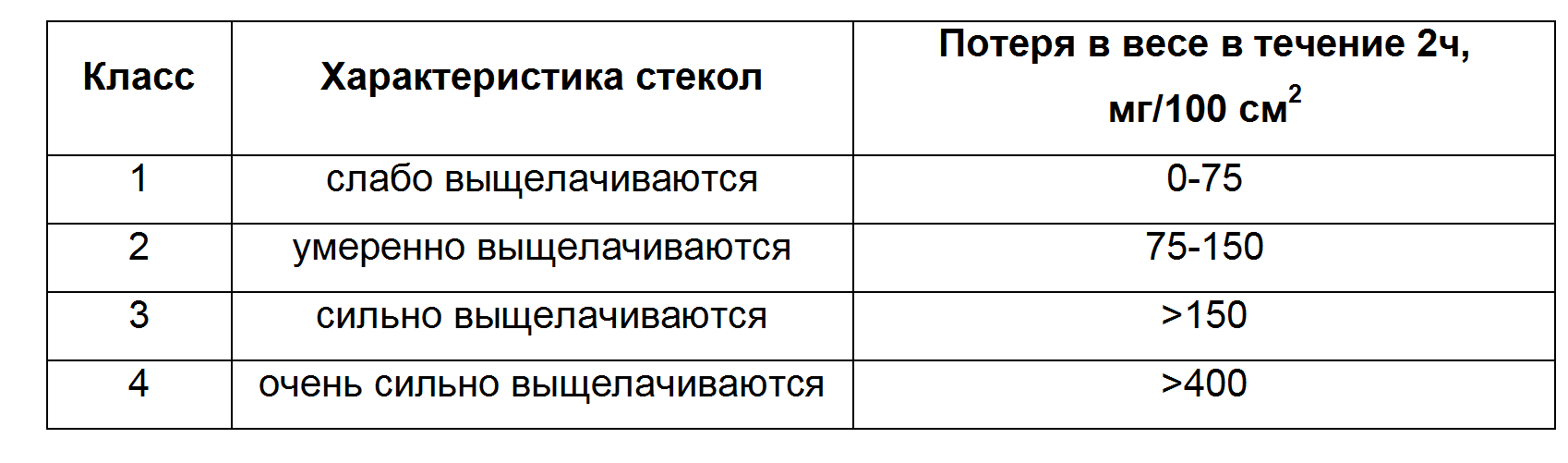
Контрольные вопросы
1. Какими компонентами стекла определяется его гидролитическая устойчивость?
2. Поясните сущность метода определения гидролитической устойчивости стекол.
3. Какими компонентами стекла определяется его устойчивость к кислотам?.
4. Поясните сущность метода определения устойчивости стекол к кислотам.
5. Какими компонентами стекла определяется устойчивость к щелочам?.
6. Знаете ли Вы сущность метода получения кварцевого стекла из боросиликатного стекла?
Лабораторная работы №5
АНАЛИЗ ТРОЙНОГО КАРБОНАТА [3] (Продолжительность лабораторной работы 6 ч; домашняя подготовка 4 ч) ЗАДАНИЕ К РАБОТЕ 1. Определение углекислого кальция. 2. Определение углекислого бария. 3. Определение углекислого стронция. УКАЗАНИЯ К РАБОТЕ 1. Определение углекислого кальция. Сущность метода Определение кальция в тройном карбонате основано на отделении кальция от стронция и бария путем обработки азотнокислых солей этих металлов спиртоэфирной смесью. При этом азотнокислый кальций вследствие хорошей растворимости в смеси спирта и эфира переходит в раствор; азотнокислые соли бария и стронция практически нерастворимы в спиртоэфирной смеси и остаются в осадке. Перевод карбонатов в нитраты достигается обработкой пробы азотной кислотой. Определение кальция заканчивается весовым методом, осаждением оксалатом аммония и прокаливанием оксалата кальция до окиси кальция. Необходимые реактивы и растворы: Кислота азотная, ч.д.а., пл. 1,40, разбавленная 1:1 и 1:10 Спиртоэфирная смесь - смесь равных объемов абсолютизированного этилового спирта и серного эфира. Кислота соляная, ч.д.а., пл. 1,19, разбавленная 1:1. Аммоний щавелевокислый, х.ч., насыщенный 1%-ый раствор. Аммиак, ч.д.а., 25%-ый, разбавленный 1:1 Серебро азотнокислое, ч.д.а., 0,5%-ый раствор. Ход анализа 1г карбоната растворяют в фарфоровой чашке в 15мл азотной кислоты, разбавленной 1:10, раствор выпаривают при температуре 130оС на песчаной бане досуха и выдерживают при этой температуре примерно 2 часа. По охлаждении к сухим солям прибавляют 15мл спиртоэфирной смеси, растирают стеклянной палочкой и переносят в сухую колбу емкостью 250мл. Остаток в чашке смывают в ту же колбу 25мл спиртоэфирной смеси, закрывают пробкой, взбалтывают и оставляют на 12 часов. Осадок отфильтровывают на фильтр с синей лентой, смоченный спиртоэфирной смесью, промывают той же смесью (12 раз по 6мл смеси) и сохраняют для определения стронция и бария. Фильтрат, содержащий весь кальций, переносят в фарфоровую чашку, выпаривают досуха и остаток после выпаривания растворяют в воде. Полученный раствор отфильтровывают от механических примесей. В растворе осаждают кальций, для чего раствор разбавляют водой до объема 100÷150 мл, подкисляют 5мл соляной кислоты, нагревают до кипения и прибавляют 5мл кипящего насыщенного раствора щавелевокислого аммония, а затем аммиак до слабощелочной среды и осадок щавелевокислого кальция оставляют на ночь. Осадок отфильтровывают на фильтр с синей лентой, промывают 1%-ым раствором щавелевокислого аммония до удаления ионов хлора в промывных водах, слегка подсушивают на воронке и переносят во взвешенный платиновый тигель, осторожно озоляют, прокаливают в муфельной печи при температуре 1100оС и взвешивают в виде СаО. Процентное содержание карбоната кальция вычисляют по формуле.
М= ((А· F)/а) 100%, (5.1) где М - определяемый компонент; А - вес осадка,г; а - навеска пробы, взятая для определения,г;F- пересчетный фактор (стехиометрический множитель). Фактор пересчета СаО на СаСО3 равен 1,7848.2.Определение углекислого бария. Сущность метода. ВаСО3 в тройном карбонате определяют из раствора азотнокислых солей бария и стронция (после отделения кальция) осаждением в виде хромата бария в слабоуксуснокислой среде. При этом происходит количественное отделение бария от стронция, поскольку хромат стронция в отличие от хромата бария в разбавленных растворах не осаждается. ОпределениеВаСО3 заканчивается весовым методом - осадок хромата прокаливают и взвешивают.Необходимые реактивы и растворы: 1. Кислота уксусная (ледяная), ч.д.а., разбавленная 1:3. 2. Аммоний хромовокислый, ч.д.а., 10%-ый раствор. 3. Аммоний уксуснокислый, ч.д.а., 3%-ый раствор. 4. Кислота азотная, ч.д.а., пл.1,4, разбавленная 1:10. 5. Аммиак, ч.д.а, 25%-ый, разбавленный 1: 2. 6. Кислота соляная, ч.д.а, пл.1,19, разбавленная 1:1. 7. Натрий серноватистокислый, ч.д.а., 0,05н. раствор.Ход анализа Осадок на фильтре (после отделения кальция спиртоэфирной смесью), состоящий из азотнокислого бария и стронция, растворяют в небольшом количестве горячей воды, доводят объем до 200-250мл, прибавляют 5÷7мл разбавленной 1:3 уксусной кислоты, раствор нейтрализуют аммиаком до слабо-кислой реакции (по лакмусовой бумажке), приливают при помешивании 70мл 10%-ного раствора хромовокислого аммония и оставляют в теплом месте на 1,5-2 часа. После отстаивания раствор фильтруют через фильтр с синей лентой, осадок в стакане и на фильтре промывают декантацией теплым 3%-ым раствором уксуснокислого аммония, полноту отмывки определяют по обесцвечиванию фильтра. Стакан в котором проводилось осаждение, ставят под воронку, основной осадок смывают струей воды в стакан (осторожно прорывая отверстие в фильтре), оставшийся на фильтре осадок растворяют небольшим количеством горячей азотной кислоты (1:10), добавляемой каплями по стенкам фильтра, и, наконец, фильтр промывают 2-3 раза водой; затем медленно при постоянном помешивании прибавляют аммиак до слабоаммиачной среды, после чего раствор вновь нейтрализуют уксусной кислотой до слабокислой среды (по лакмусовой бумажке). Разбавляют раствор до объема 100÷150 мл, нагревают до кипения, прибавляют 10мл 10%-ного раствора хромовокислого аммония и дают отстояться в теплом месте 1,5-2 часа. Осадок отфильтровывают через два фильтра с синей лентой, промывают сначала 3-4 раза декантацией, а затем на фильтре 3%-ным раствором уксуснокислого аммония до обесцвечивания фильтра. Фильтраты и промывные воды после обоих фильтрований осадка хромата бария объединяют вместе и сохраняют для определения стронция. Осадок дополнительно промывают 2 раза теплой водой, подсушивают на воронке в сушильном шкафу, фильтр осторожно озоляют при свободном доступе воздуха, а осадок прокаливают в муфельной печи при 800оС. Процентное содержаниеВаСО3 вычисляют по формуле 5.1, фактор пересчета ВаСrCO3 наВаСО3 равен 0,7790.3. Определение углекислого стронция. Сущность метода. В фильтре после осаждения бария стронций выделяют в виде карбоната стронция, последний растворяют в соляной кислоте и осаждают в виде сернокислой солиSrSO4. Относительная ошибка определения 0,5%. Необходимые реактивы и растворы: 1. Раствор углекислого аммония: 55г углекислого аммония, х.ч., растворяют в 200мл воды, добавляют 24мл 25%-ного аммиака, ч.д.а, и перемешивают. 2. Кислота соляная, ч.д.а, пл.1,19, разбавленная 1:5. 3. Кислота серная, ч.д.а, пл.1,84, разбавленная 1:3. 4. Спирт этиловый, 90%-ый и разбавленный 1:1. 5. Барий хлористый, ч.д.а., 10%-ый раствор.Ход анализа Фильтраты после весового определенияВаСО3 (после 1-го и 2-го осаждений) соединяют вместе, выпаривают до объема 150÷200мл, нагревают до кипения, прибавляют 30÷40мл раствора углекислого аммония и раствор с осадком карбоната стронция оставляют в теплом месте на 1÷2 час. Затем осадок отфильтровывают на фильтр с синей лентой и промывают горячей водой до обесцвечивания фильтра. Осадок на фильтре осторожно растворяют в горячей соляной кислоте1:5), избегая разбрызгивания и промывают фильтр несколько раз горячей водой. К полученному раствору приливают при помешивании 15мл серной кислоты (1:3), добавляют этиловый спирт (для уменьшения растворимости осадкаSrSO4) в объеме, равном объему раствора, и оставляют раствор на 12 час. Осадок отфильтровывают на фильтр с синей лентой, промывают спиртом, разбавленным 1:1, содержащим несколько капель серной кислоты, а затем 96%-ым спиртом до удаления серной кислоты (проба с хлористым барием). Осадок подсушивают, осторожно озоляют и прокаливают в муфельной печи при темно-красном калении до постоянного веса. Процентное содержаниеSrCO3 вычисляют по формуле 5.1, фактор пересчетаSrSO4 наSrCO3 равен 0,7526. Контрольные вопросы:
1. Каким образом из тройного карбоната получается оксидный эмиттер электронов?
2. Поясните сущность метода определения количества углекислого кальция в тройном карбонате.
3. Поясните сущность метода определения количества углекислого бария в тройном карбонате.
4. Поясните сущность метода определения количества углекислого стронция в тройном карбонате.
5. Почему в качестве эмиссионного покрытия используется окислы металлов, а не металлы в компактном состоянии?
6. Эмиссионное покрытие из окислов щелочиоземльных металлов является полупроводником или диэлектриком?
Лабораторная
работы №6
ВЫТРАВЛИВАНИЕ
МОЛИБДЕНОВОГО И
СТАЛЬНОГО КЕРНА
(Продолжительность
лабораторной работы 4 ч; домашняя
подготовка 2 ч)
Сущность
метода.
Для
изготовления точных спиралей из тонких
вольфрамовых проволок они навиваются
на точно калиброванную молибденовую
или стальную проволоку (керн). Такие
керны допускают применение высокой
температуры при формовании спирали,
которое осуществляется или в печах с
защитной атмосферой в молибденовых
лодочках или прямым накаливанием керна
электрическим током в водороде. Керн
впоследствии удаляется растворением
его в кислотах. Спираль после вытравливания
керна, промывки и сушки не должна быть
деформирована.
ЗАДАНИЯ К РАБОТЕ
1.
Приготовление раствора для вытравливания
молибденового керна.
2. Приготовление
раствора для вытравливания стального
керна.
3. Вытравливание кернов.
Необходимые
реактивы и растворы:
1.
Вода дистиллированная.
2. Кислота
азотная.
3. Кислота серная.
4. Кислота
соляная.
5.
Индикатор
метиловый оранжевый или фенолфталеин.
6. Натрий или калий гидроокись.
7.
Ацетон.
1.
Приготовление раствора для вытравливания
молибденового керна.
Количество
кислот и
воды для
приготовления раствора рассчитывается
по формулам:

где
V1,
V2,
V3,
V4
-
объемы
исходных азотной и серной кислот, воды
и приготавливаемого раствора, л; с1
и c2
-
концентрация
исходной азотной и серной кислот,
массовые проценты; d1
и d2
- плотность
исходной азотной и серной кислот при
температуре 20oС;
t1
и t2
-
температура
азотной и серной кислот, оС;
54,80 и 27,40 -
произведение
концентраций азотной (40) и серной (20)
кислот в массовых процентах в данной
смеси на плотность смеси .
2.
Приготовление раствора для вытравливания
стального керна
Количество
кислот и воды, необходимое для
приготовления вытравливаемого раствора
рассчитывается по формулам:

где V1, V2, V3, V4 - объемы исходных азотной и серной кислот, воды и приготавливаемого раствора, л; с1 и c2 - концентрация исходной азотной и серной кислот, массовые проценты; d1 и d2 - плотность исходной азотной и серной кислот при температуре 20oС; t1 и t2 - температура азотной и серной кислот, оС; 25,20 - произведение концентраций азотной (40) и серной (20) кислот в массовых процентах в данной смеси на плотность смеси . Приготовление вытравливающих растворов производить в емкостях из химически и термически стойких материалов. При этом осторожно вливать в воду азотную, а затем серную кислоту. Затем раствор перемешать и охладить до комнатной температуры. Щелочной раствор готовят в емкостях из винипласта, применяя дистиллированную воду. Наименование операций, реактивов и их характеристик, температуры и продолжительности операций для спиралей с молибденовым керном приведены в табл. 6.1. Таблица 6.1


Наименование операций, реактивов и их характеристик, температуры и продолжительности операций для спиралей со стальным керном приведены в таблице 6.2. Таблица 6.2

Контроль качества:
1. Операции вытравливания керна производить без применения внешнего нагрева до прекращения выделения бурых паров окислов азота. 2. Запрещается оставлять в вытравливающем растворе спирали без керна и мокрые спирали на воздухе. 3. Нейтральность промывных вод после промывки спирали вытравливающего раствора проверять метиловым оранжевым, после промывки их от щелочи фенолфталеином. 4. Спирали при вытравливании, очистке, промывке и обезвоживании должны полностью погружаться в соответствующую жидкость. 5. Спирали после сушки должны иметь чистую, без деформации поверхность.Контрольные вопросы:1. Поясните конструкцию биспиральных и триспиральных конструкций электродов люминесцентных ламп и технологию их изготовления.
2. Опишите функции керна из молибденовой или стальной проволоки при изготовлении спиралей, биспиралей и триспиралей.
3. Какова сущность метода вытравливания молибденового керна?
4. Какова сущность метода вытравливания стального керна?
5. Каковы основные правила работы с растворами для вытравливания молибденового и стального кернов?
6. Как действуют на вольфрамовую спираль растворы для вытравлива- ния кернов?
7. Что Вы знаете об утилизации молибденосодержащего раствора (после вытравливания керна)?
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ К ВЫПОЛНЕНИЮ ЛАБОРАТОРНЫХ РАБОТ Количество лабораторных работ, выполняемых студентами, определяется числом часов по учебному плану специальности. Лабораторные работы по дисциплине «Химия материалов со специальными свойствами» выполняются группой из 2-3 человек. Преподаватель заранее, в начале семестра, сообщает последовательность выполнения лабораторных работ, поэтому на занятия студенты должны приходить, ознакомившись с теоретическим материалом по предстоящей работе. Перед началом работы преподаватель спрашивает о целях и задачах работы, затем после краткого инструктажа, студенты приступают к выполнению работы. В конце занятия преподаватель проверяет полученные результаты и делает отметку в журнале. По выполненной лабораторной работе каждым студентом составляется отчет в соответствии с требованиями СТО МордГУ 013-2003 «Работы лабораторные. Правила оформления отчетов», допуск к следующей работе студент получает после сдачи отчета о предыдущей лабораторной работе. Сдача работ проводится в конце занятия, либо в дополнительно назначенное время и предусматривает проверку результатов проведенных измерений, исследований и расчетов, правильность оформления отчета и ответы на контрольные вопросы. Защищенную работу преподаватель передает на кафедру, где она хранится в течение семестра.
ЛИТЕРАТУРА
1. Эспе В. Технология электровакуумных материалов: том 1. Металлы и материалы с металлической проводимостью. – М.-Л.: Госэнергоиздат. – 1962. – 632 с.; том 2. Силикатные материалы. – М.: Энергия. – 1968. – 448 с.; том 3. Вспомогательные материалы. – М.-Л.: Энергия. – 1969. – 368 с.
2. Чуркина Н.И., Литюшкин В.В., Сивко А.Н. Основы технологии электрических источников света. – Саранск.: Морд. кн. изд-во, 2003. – 344 с.
3. Кличко Ю.А. Методы анализа материалов, применяемых в электровакуумной промышленности. – М.: Советское радио. – 1972. – 408 с.
4. Гавзе М.Н. Взаимодействие ртути с металлами и сплавами. – М.: Наука. – 1966. – 160 с.
5. Гугель Б.М. Люминофоры для электровакуумной промышленности. – М.: Энергия. – 1967. – 344 с.
6. Охонская Е.В., Федоренко А.С. Расчеты и конструирование люминесцентных ламп: Учеб. – Саранск: Изд-во Мордов. ун-та. – 1997. – 184 с.
7. Федоренко А.С. Люминесцентные лампы (Расчёт, моделирование, экспериментальные исследования, создание конструкторских и технологических решений) / А.С. Федоренко. – Саранск: Изд-во СВМО, 2009.- 333 с.
8. Айзенберг Ю.А. Основы конструирования световых приборов: Учебное пособие для вузов. – Энергоатомиздат, 1996. – 704 с.
9. Вредные химические вещества. Неорганические соединения элементов I-IVгрупп: Справ. изд./ Под ред. В. А. Филова и др.: Химия, 1988. – 512 с.
10. Принсгейм Л. Флуоресценция и фосфоресценция. ИЛ. 1951.
11. Девятых Э.В. Люминесцентные лампы. Люминофоры и люминофорные покрытия / Э.В.Девятых, В.Ф.Дадонов – Саранск: Изд-во Мордов. ун-та, 2007, - 344 с.
12. Каталог ИСМАН.
13. Мишенина Л.Н. Структуро- и фазообразование люминофоров на основе оксосульфидов РЗЭ и сложных алюминатов ЩЗМ в процессах СВС, автореферат дисс. кандидата химических наук, Томский госуниверситет, Томск, 1996г.
14. А.с. 1467983 СССР, МКИ4С09К 11/59, 11/83. Люминесцентный состав для газоразрядных ламп высокого давления / Б.В.Черновец, Э.В. Девятых, Р.И. Хра- мова, А.С.Федоренко и др.
15. Ульмишек Л.Г. Производство электрических ламп накаливания. Изд-во «Энергия», М.-Л., 1966, 640 с.
16. Дубок В.А., Пронькин В.С. Массоперенос и диффузия примесей в поликоровой керамике натриевых ламп высокого давления.- В кн.: Электрические источники света (Труды ВНИИИС, вып.10), Саранск, Мордов. кн. изд-во, 1978, с. 123-129.
17. Дэшман С. Научные основы вакуумной техники. Изд-во «МИР», М., 1964, 716 с.
18. Патент № 2078316 Российская Федерация МКИ6G01K5/22. Прибор для измерения и регулирования температуры / А.С.Федоренко, Л.М.Лавренко, А.А.Прытков, В.А.Горюнов, Д.А.Федоренко – Опубл. 27.09.97, Бюл. №12.
19. Микаева С.А. Создание нового поколения люминесцентных устройств с улучшенными световыми характеристиками. Издательство «Научтехлитиздат», - Москва, 2004.- 210 с.
20. Паншин Ю.А., Малкевич С.Г., Дунаевская Ц.С. Фторопласты. Л: «Химия», 1978, 215 с.
21. В.Г.Сидоров, А.С.Федоренко, Внешние композиционные светотехнические покрытия на колбах источников света, «Светотехника», 2001, №6, С.20-22.
22. Патент № 2079774 Российская Федерация МКИ6 F21V9/10. Композиционный светотехнический материал / В.А.Горюнов, Л.М.Лавренко, А,С.Федоренко, Б.Н.Денисов, Е.В.Никишин, Д.А.Федоренко, В.Я.Гришаев, Р.А.Федоренко – Опубл. 27.11.2001, Бюл. № 33.
23. Патент № 2065639 Российская Федерация МКИ6H01J61/35, 61/40, 61/44. Источник света /А.С.Федоренко, Д.А.Федоренко, Р.А.Федоренко, Л.М.Лавренко, В.А.Горюнов – Опубл. 20.04.96, Бюл. № 23.
24. Куприянов И.П. Технологический микроклимат. М., «Сов. радио», 1976, 176 с.
25. Таиров Ю.М., Цветков В.Ф. Технология полупроводниковых и диэлектрических материалов. Учеб. для вузов. – 3-е изд. стериот. – Санкт-Петербург.: Из-во “Лань”, 2002. – 424 с.
26. Курносов А.И., Юдин В.В. Технология производства полупроводниковых приборов и интегральных микросхем. 2-е изд. – М.: Высшая школа. – 1979. – 367 с.
27. Коган Л.М. Светодиоды нового поколения для светосигнальных и осветительных приборов. Новости светотехники. Выпуск 7-8 (34-35)//Под редакцией Ю.Б. Айзенберга. – М: Дом света. – 2001. – 47 с.
Оглавление
стр.
А. ХИМИЯ МАТЕРИАЛОВ ДЛЯ ИСТОЧНИКОВ СВЕТА
И ЭЛЕКТРОВАКУУМНЫХ ПРИБОРОВ 3
1. Принципы выбора и методы обработки материалов
со специальными свойствами 3
2. Металлы 4
2.1. Тугоплавкие металлы 4
2.1.1. Вольфрам 5
2.1.2. Молибден 7
2.1.3. Ниобий 8
2.2.Никель 9
2.3.Медь 9 2.4. Платинит 11
2.5. Щелочные металлы 12
2.6. Ртуть 12
2.7. Амальгамы 15
3. Газопоглотители 17
4. Люминесценция, люминофоры и покрытия 24
4.1. Определение, виды, законы, характеристики 24
4.2. Основы технологии изготовления люминофоров 32
4.3. СВС-технология изготовления люминофоров 37
4.4. Люминофоры для компактных, ультрафиолетовых ламп
и ламп ДРЛ 39
4.5. Нанесение люминофорных покрытий 40
5. Химия стекла и покрытий на стекле 41
5.1. Определение и классификация стёкол 41
5.2 Химическая устойчивость стёкол 44
5.3. Обработка поверхности стекла 45
5.4. Нанесение покрытий на стекло 46
5.5. Химическое травление поверхности стекла 51
5.6. Нанесение покрытий на ЛОН 53
6. Кварцевое стекло (КС) 56
7. Керамика 58
8. Ситаллы 61
9. Газы и пары металлов 62
10. Эмиссионные покрытия для электродов 64
11. Припои для соединения деталей приборов 67
12. Эластомеры и полимеры 69
13. Вакуумные уплотнители и смазочные материалы,
органические рабочие жидкости 72
14. Цоколёвочные мастики 74
Б. ХИМИЯ МАТЕРИАЛОВ ДЛЯ СВЕТОДИОДОВ
И ПОЛУПРОВОДНИКОВЫХ ПРИБОРОВ 75
15. Электропроводность полупроводников 75
16. Полупроводники (общие сведения и классификация). 77
17. Химические процессы технологии материалов электронной техники 81 18. Обработка материалов 86
19. Химические процессы фотолитографии 86
20. Химические процессы при эпитаксии. 89
21.Получение защитных плёнок 91
22. Диффузия и ионная имплантация (для соединений AIIIBV) 92
23. Травление полупроводников 93
24. Получение деионизованной воды 93
25. Химия металлов и металлических плёнок 94
Лабораторная работа № 1 95
Лабораторная работа № 2 97 Лабораторная работа № 3 98
Лабораторная работа № 4 100
Лабораторная работа № 5 102
Лабораторная работа № 6 106
Методические рекомендации к кыполнению лабораторных работ 109
Литература 109
Оглавление 111