

Борман Физическая кинетика атомных процессов в наноструктурах 2011
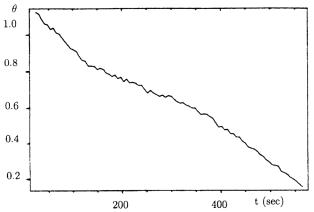
.pdfРис. 1.5. Зависимость степени покрытия поверхности Si(100) кислородом от времени экспозиции при давлении p = 1×10–6 Тор и температуре:
1 − 970 К, 2 − 1050 К, 3 − 1110 К, 4 − 1150 К, 5 −1190 К
Вблизи порога окисления при увеличении температуры увеличиваются время tc и кри-
тическая концентрация хемсорбированного кислородаθс ,
при достижении которых происходит переключение режима хемосорбции на режим образования оксида. Такие зависимости tc (T ) и θc (T ) свидетельст-
вуют о возможных каналах ухода атомов кислорода с поверхности.
Как было отмечено выше, в процессе вакуумного отжига происходит уменьшение поверхностной концентрации ки-
τок, с
Рис. 1.6. Зависимость характерного
времени окисления τок от температуры при Р = 6 10–7 Тор
41
слорода. Проведенные дополнительные эксперименты показали, что отжиг в течение 10 мин приводит к уменьшению на 90 % концентрации поверхностного кислорода (рис. 1.6). Сравнение кинетических зависимостей, представленных на рис. 1.5 и 1.6 показывает, что характерное время ухода кислорода с поверхности при температурах T = 1020 К сравнимо со временем формирования оксида SiO2 (t = 500 c). Это означает, что при анализе начального окисления кремния необходимо учитывать процесс ухода кислорода с поверхности.
Рис. 1.7. Кинетика изменения концентрации кислорода в процессе вакуумного отжига при Т = 1020 К в течение 10 мин
Полученный результат является показателем преимущества метода измерения кинетики в режиме реального времени, особенно при высоких температурах. Имеются два возможных канала ухода кислорода с поверхности: диффузия в объем кристалла и десорбция в виде SiO [22]. Из массоспектрометрического анализа [23] следует, что сам кислород в виде атомов и молекул не десорбируется вплоть до 1400 К. Провести различие между каналами ухода кислорода на основе результатов, полученных в описанных экспериментах, не представляется возможным. Появление дополнительного пика O1s с Есв = 532,4 эB (см. рис. 1.2,в) означает, что имеет место диффузия атомов кислорода в объем кристалла. Что касается канала десорбции, то известно, что SiO десорбируется в диапазоне температуры (975–1400 К) [24], в котором и были проведены исследования.
42
На основании результатов по исследованию механизма начального роста оксида SiO2 на поверхностях Si(100) и Si(111) было установлено, что наблюдается рост оксидной фазы в виде отдельных островков, существует критические давление и температура, экспозиция [24] и критическая степень покрытия (θ = 0,3 [25]), а также сосуществуют адсорбированная и оксидная фазы. С помощью метода просвечивающей электронной микроскопии высокого разрешения в работе [36] в оксидных пленках SiO2, выращенных в атмосфере кислорода на поверхности (100), наблюдались островки размером до 10 нм.
Доказательством существования островков оксида на поверхности Si(001) являются также результаты исследований [26] с помощью РФЭС неоднородного окисления поверхности, что может свидетельствовать об образовании отдельных микроскопических фаз SiO2 (кластеров) на поверхности Si(001). Островковые пленки SiO2 были идентифицированы в работе [27] при исследовании структуры интерфейса Si/SiO2 методом фотоэлектронной спектроскопии высокого разрешения.
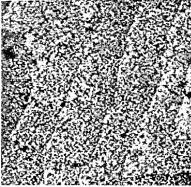
Прямым доказательством существования кластеров естественного оксида SiO2 на Si(111) могут являться результаты исследования с помощью СТМ роста оксида при комнатной температуре и
атмосферном давлении [37]. |
|
|
||||
На рис. 1.8 представлено СТМ- |
|
|
||||
изображение, |
подтверждающее |
|
|
|||
образование |
зародышей |
малых |
|
|
||
кластеров 1÷2 нм в диаметре в |
|
|
||||
верхнем слое кремния (черные |
|
|
||||
точки). Формирование кластеров |
|
|
||||
происходило |
путем |
гомогенного |
|
|
||
зародышеобразования |
на |
боль- |
|
|
||
ших временах (до 800 ч). Ввиду |
|
|
||||
того, что метод СТМ не позволяет |
|
|
||||
получать рельефы |
поверхности |
Рис. 1.8. 2 |
СТМ-изображение |
|||
диэлектрических объектов, к чис- |
||||||
лу которых |
относятся |
островки |
400*400 нм |
топографии поверх- |
||
ности Si(111) после экспозиции в |
||||||
SiO2, авторами была предложена |
кислороде в течение 400 ч в воз- |
|||||
оригинальная |
методика получе- |
духе при атмосферном давлении |
||||
ния СТМ-изображений, |
окислен- |
и температуре 300 К |
||||
43
ной на начальной стадии поверхности Si, путем визуализации кластеров SiO2 после их удаления и последующего анализа топографии поверхности кремния (дырки на месте удаленных островков оксида). Этот метод подобен методу реплик в просвечивающей электронной микроскопии.
Принимая во внимание представленные выше результаты, можно заключить, что формирование и рост оксидных островов, пороговый характер формирования оксида (т.е., существование критического покрытия, критической экспозиции и критического давления), сосуществование адсорбированной и оксидной фаз, подтверждают возможность интерпретации процесса формирования оксида как фазового перехода первого рода.
1.3. Кинетика послойного роста первых монослоев оксида SiO2
Кинетика послойного окисления исследовалась непосредственно в камере анализатора электронного спектрометра XSAM-800 в режиме реального времени [24] в отличие от обычно используемого режима измерения по точкам после определенной экспозиции [25, 26]. При напуске кислорода в камеру (р = 1×10–6Тор) регистрировались Si2p и O1s спектры один за другим каждые 3 мин. Температура образца 1135 К выбиралась в соответствии с общепринятым режимом окисления кремния [27, 28]. Спектры Si2p после окисления раскладывались на компоненты, соответствующие характерным стадиям окисления.
На рис. 1.9 представлена кинетика накопления атомов кремния в различных состояниях Sin+ в процессе окисления. Для наглядности зависимостей просуммированы концентрации атомов Si в состояниях Si1+ Si2+ (линия Si12), соответствующих двум хемосорбированным состояниям кислорода, и в состояниях Si3+ Si4+ (линия Si34), соответствующих кремнию в SiO2. Суммарная концентрация всех четырех состояний (линия Si∑) соответствует количеству атомов Si с разрушенной связью Si–Si окруженных разным количеством атомов кислорода. Интенсивность линии Si∑ равна площади, полученной после вычитания площади соответствующей пику Si2p подложки из общей площади под всей линией спектра, т.е. пло-
44
щадь ограниченная плечом (см. рис. 1.3). В данном случае следует
подчеркнуть, что интенсивность Si∑ не зависит от положения пиков Si1+, Si2+, Si3+ и Si4+. Из рис. 1.9 видно, что линия Si∑ имеет три ступени по интегральной интенсивности фотоэлектронов Si2p, равных соответственно, 100, 200 и 300 условных единиц. Для определения концентрации атомов Si, соответствующих этим интенсивностям, можно использовать тот факт, что чистой поверхности кремния отвечает интенсивность ISi = 2000 условных единиц. Так как геометрия эксперимента такова, что направление покидающих поверхность фотоэлектронов перпендикулярно к поверхности, величина ISi определяется уравнением
ISi = ∫0∞ I0 exp(−z/λSi)dz = I0λ Si ,
где z – расстояние от поверхности, λSi – средняя длина свободного побега фотоэлектронов в кремнии (для излучения MgKα hν = = 1253,6 эВ, λSi = 1,3 нм) и I0 – интенсивность поверхностных атомов кремния на единице длины. Принимая во внимание, что величина межплоскостного расстояния для Si(100) равна 1,3 А, легко оценить, что сигнал Si2p подложки (ISi) определяют 10 слоев кремния. Таким образом, один поверхностный слой в кремнии дает вклад (если пренебречь экспоненциальным ослаблением) в сигнал интенсивностью примерно равной 200 условных единиц. Также необходимо отметить, что концентрация атомов в кремнии 5×1022 см–3, что примерно в два раза больше, чем в двуокиси кремния (2,3×1022 см–3). Поэтому одному слою оксида соответствует интенсивность сигнала Si2р фотоэлектронов 100 условных единиц. Именно такой интенсивности соответствует высота ступени на рис. 1.9. Таким образом, можно заключить, что ступени на линии Si∑ являются результатом последовательного формирования оксидных слоев на поверхности Si(100), и каждая из этих трех ступеней соответствует одному монослою. Толщина окисленной пленки при Т = 1135 К в течение 60 мин дает соответствие трем монослоям окисла (см. рис. 1.9). Это количество слоев хорошо согласуется с величиной отдельно полученной с помощью калибровки интенсивности O1s фотоэлектронов.
45
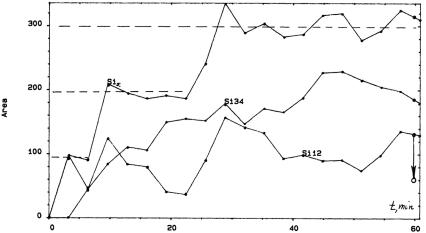
Рис. 1.9. Кинетика накопления атомов кремния в различных оксидных состояниях в процессе начального окисления (сплошная линия). Стрелка справа показывает, что Si1+ и Si2+ состояния уменьшаются в результате последующего вакуумного отжига. Кривая Si∑ соответствует концентра-
ции атомов кислорода во всех четырех оксидных состояниях, т.е. сумме
Si1+, Si2+, Si3+ и Si4+ состояний
Рассмотрим теперь распределение атомов кремния между разными состояниями в процессе окисления, сравнивая Si12, Si23 и Si∑ линии на рис. 1.9. На начальном этапе (t < 3 мин) Si3+и Si4+ не появляются. Следовательно, увеличение сигнала Si∑ обусловлено исключительно появлением и накоплением Si1+и Si2+ состояний. Кроме того, в течение периода времени от 3 до 6 мин количество атомов кремния во всех оксидных состояниях (Si∑) остается почти постоянным и соответствует одному монослою оксида. Одновременно линия Si12 опущена вниз, в то время как линия Si34 поднимается. Другими словами, имеет место переход атомов кремния из состояний Si1+, Si2+ в состояния Si3+ и Si4+, который завершает формирование монослоя оксида. В процессе увеличения концентрации Si∑ до величины второй ступени, соответствующей двум слоям окисла (6 < t < 9 мин), снова происходит накопление состояний Si1+, Si2+. На этапе t = 9–21 мин эти состояния снова трансформируются в Si3+и Si4+. Подобный результат наблюдается также на третьем этапе (т.е. накопление Si1+, Si2+ состояний при t = 21–27
46
мин и их трансформация в состояния Si3+и Si4+ при t = 27–60 мин). После 60 мин окисления (конец третьего этапа) количество Si1+, Si2+ состояний остается значительным по сравнению с Si3+и Si4+. Однако вакуумный отжиг (р = 10–9 Тор) при Т = 1135 К в течение 15 мин приводит примерно к двукратному уменьшению концентрации атомов кремния в состояниях Si1+, Si2+ (смотри стрелку на рис. 1.9). Заслуживает внимания тот факт, что это уменьшение обусловлено значительным уменьшением Si1+-состояния, в то время как Si2+ изменяется в пределах 15 %.
Таким образом, можно сделать вывод, что формирование монослоя оксида сопровождается накоплением Si1+- и Si2+-состояний с последующим переходом их в состояния Si3+, Si4+ при неизменном суммарном количестве атомов кремния во всех Sin+ состояниях (Si∑ линия). Такое поведение характерно для роста каждого из трех оксидных слоев, в том числе и для первого монослоя окисла, где существование ступени при 3–6 мин менее убедительно, поскольку зафиксировано только две точки на рис. 1.9.
На основе полученных экспериментальных данных можно предложить следующий механизм формирования оксидного слоя на начальных стадиях термического окисления кремния. Рост каждого слоя может быть разделен на две стадии. На первой происходит накопление атомов кислорода на границе раздела. Рост диоксида кремния, как было установлено, происходит путем диффузии кислорода. Природа диффузии зависит от толщины оксидной плен-
ки [37].
Для тонких пленок (2÷5 нм) было показано, что механизм роста оксида отличен от механизма роста толстых пленок. Это различие обусловлено транспортом атомов кислорода через оксид. В обоих случаях диффузия атомов кремния не лимитирует рост оксида. Что касается сверхтонких пленок (< 1 нм), роль транспорта атомов кремния и кислорода до конца не ясна в роли накопления их на границе раздела Si/SiO2. Это накопление сопровождается появлением состояний Si1+, Si2+. Результаты экспериментов, проведенных на оксидных пленках толщиной 1÷5 нм, подтвердили локализацию Si1+- и Si2+-состояний в узкой области (от 0,2 нм до 0,6÷1 нм) вблизи границы раздела. На второй стадии при достижении на интерфейсе определенной концентрации кислорода (или атомов кремния в Si1+- и Si2+-состояниях) происходит фазовый переход первого ро-
47
да. Это приводит к появлению атомов кремния в Si3+-, Si4+-со- стояниях и к уменьшению состояний Si1+, Si2+. В соответствии с [15, 32, 37] формирование поверхностного оксида часто сопровождается структурным переходом в поверхностном слое подложки. Для образования новой фазы (оксид) требуется определенное время фазового перехода (τрт). Следовательно, в ходе эксперимента, проводимого в режиме реального времени, из-за короткого времени измерения (τэкс) по сравнению с (τрт), фазовый переход может не завершиться. В результате концентрация Si1+, Si2+ не падает до нуля (см. рис. 1.9) в пределах временных интервалов соответствующих этапам, в частности, третьему этапу. Совершенно другая ситуация наблюдается после вакуумного отжига. При таком отжиге времена (τэкс) и (τрт) по-видимому становятся сравнимыми, что приводит к
уменьшению состояний Si1+, Si2+ наблюдаемому в эксперименте (стрелка на рис. 1.9). Переход Si1+, Si2+ в Si3+, Si4+ в результате от-
жига также наблюдался в [27].
Контрольные вопросы к главе 1
1.Дайте физическую интерпретацию наличия в РФЭ спектре Si2p пленки оксида кремния состояний Sin+ (n = 1, 2, 3, 4).
2.Объясните методику определения кислорода на поверхности
Si(001).
3.Проанализируйте зависимость начальной скорости окисления кремния от температуры (уменьшение скорости с ростом Т).
4.Укажите, на основании каких признаков окисление кремния можно представить как фазовый переход первого рода?
5.Прокомментируйте механизм послойного роста оксида SiO2 на поверхности Si(001) (~ 3 монослоя).
6.Используя Р–Т диаграмму окисления поверхности кремния Si(001) опишите режимы пассивного, активного окисления, а также переходный режим.
7.Объясните наличие критического времени (времени отсечки),
при котором достигается значение θC степени покрытия поверхно-
сти Si(001) кислородом, необходимое для начала роста SiO2 при данных давлении и температуре.
48
Список литературы
1.Carriere В., Chouiyakh A. and Lang В. Surf. Sci. 126, 495 (1983).
2.Pianetta P., Lindau I., Garner С.M. and Spicer W.E. Phys. Rev. Lett. 37, 1166 (1976).
3.Borman V.D., Gusev E.P., Lebedinski Yu.Yu. and Troyan V.I. Phys. Rev. В. 49, 5415 (1994).
4.Lutz F., Bischoff J.L., Kubler L. and Bolmont D. Phys. Rev. В 40, 10356 (1989).
5.Hochella M.F. Jr. and Carim A.H. Surf. Sci. 197. L260 (1988).
6.Scofield J.H. Electron Spectrosc. Relat. Phenom. 8, 129 (1976).
7.Борман В.Д., Гусев Е.П., Девятко Ю.Н., Лебединский Ю.Ю., Рогожкин С.В., Тронин В.Н., Троян В.И. Поверхность, 8, 22 (1990).
8.Lutz F., Kubler L., Bischoff J.L. and Bolmont D. Phys. Rev. В 40, 11747 (1989).
9.Hollinger G. and Himpsel F.J. Phys. Rev. В 28, 3651 (1983).
10.Himpsel F.J., McFeely F.R., Taleb-lbrahimi A., Yarmoff J.A. and Hollinger G. Phys.Rev, B38, 6084 (1988).
11.Tabe M., Chiang Т.Т., Lindau I. and Spicer W.E. Phys. Rev. B34, 2706
(1986).
12.Ibach H., Bruchmann H.D. and Wagner H. Appl. Phys. A 29, 113
(1982).
13.Grunthaner P.J., Hecht M.H., Grunthaner F.J. and Johnson N.M. Appl. Phys. 61, 629 (1987).
14.Dreiner S., Schurmann M., Westphal C. J. Elec. Spec. and Rel. Phen. 137-140 (2004) 79.
15.Hollinger G., Morar J.F., Himpsel F.J., Hughes G. and Jordan J.L. Surf. Sci. 168, 609 (1986).
16.Morgen P., Hofer U., Wurth W. and Umbach E. Phys. Rev. B39, 3720
(1989).
17.Namiki A., Tanimoto К., Nakamura Т., Ontake N. and Suzaki Т. Surf. Sci. 222, 530 (1989).
18.Stavola M., Patel J.R., Kimerling L.С. and Freeland P.E., Appl. Phys. Lett. 42, 73 (1983).
19.Tong Lee S. and Nichols D. Appl. Phys. Lett. 47,1001 (1985).
20.Mikkelsen J.С. Jr., Appl. Phys. Lett. 40. 336 (1982).
21.D'Evelyn M.P., Nelson M.M. and Engel Т. Surf. Sci. 186. 75 (1987).
22.Smith F.W. and Ghidini G. J. Electrochem. Soc. 129,1300 (1982).
23.Leibsle F.M., Samsavar A. and Chiang Т.С., Phys. Rev. В 38, 5780
(1988).
24. Борман В.Д., Гусев Е.П., Лебединский Ю.Ю., Попов А.П.,
Троян В.И. // ЖЭТФ, Т.95, Вып. 4, 1989.
49
25.Tabe M., Chiang Т.Т., Lindau I. and Spicer W.E., Phys. Rev. B34, 2706
(1986).
26.Lutz F., Bischoff J.L., Kubler L. and Bolmont D., Phys. Rev. В 40, 10356 (1989).
27.Claeys C.L., De Keermaecker R.F. and Declerck G.J. In the Si-SiО, System, edited by P. Balk (Elsevier, New York, 1988).
28.Atkinson A. Rev. Mod. Phys. 57, 437 (l985).
27. Hattori T. and Suzuki T., Appl. Phys. Lett. 43, 470 (1983)
30.Ishizaka A. and Iwata S., Appl. Phys. Lett. 36, 71 (1980).
31.Aoto N., Ikawa E., Endo N. and Kurogi Y. Surf. Sci. 243, 121 (1990).
32.Gusev E.P. and Popov A.P. Surf. Sci. 248, 241(1991).
33.Ourmazd A., Taylor D.W., Rentschler J.A. and Bevk J. Phys. Rev. Lett. 59, 2l3 (l989).
34.Ourmazd A., Fouss P.H., Bevk J. and Morar J.F., Appl. Surf. Sci. 41/42, 365 (1989).
35.Emelvanou A.V. Электрон. Промышленность 8, 36 (l983).
36.Chu A.X. and Fowler W.B. Phys. Rev. B 41, 5061 (1990).
37.Neuwald U. et. al Appl. Phys. Lett 60, (1992) 1307.
50