

Ocherki_klinicheskoy_biokhimii
.pdf281
16.4.2. Ламинин и гломерулопатии
Ламинин как причина различных заболеваний является куда интереснее, чем кажется на первый взгляд. И его структура гораздо сложнее. Известно, что он является комбинацией А, В и гамма – цепей, каждой из который существует ещё по несколько видов: 5 видов А – цепи, 4 вида В – цепи и 3 типа гамма –цепи, итого
12 видов и 12 генов, кодирующих ламинин. Разобраться во всех этих типах крайне сложно, так как разные виды ламинина обусловливают отдельное заболевание,
если что – то в их синтезе идёт не так.
Так, Ламинин-521, состоящий из а5, В2 и гамма - 1 цепей, является одним из важнейшихгликопротеинов гломерулярной базальной мембраны и играет важную
(https://journal.nephrolog.ru/jour/article/view/201)
роль в ее структуре и функции. Ламинин – 521 обеспечивает стабильность гломерулярной базальной мембраны путём одновременного связывания с интегринами эндотелиальных клеток, выстилающих почечные сосуды клубочков изнутри и коллаген IV типа, и тем самым поддерживая селективность гломерулярной мембраны в отношении фильтрации и образования первичной мочи. Роль ламинина особенно заметна в случае генетических дефектов.
Аутосомно-рецессивная мутация гена LAMB2, кодирующего синтез β2 - цепи ламинина, сопровождается развитием врожденного нефротического синдрома с офтальмологическими и неврологическими расстройствами, известного как
синдром Пирсона.
Нефротический синдром характеризуется нарушением селективности гломерулярной мембраны почечных клубочков, которое выражается в потери большого количества белка, преимущественно альбумина, с последующей гипопротеинемией.
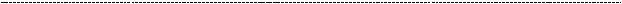
282
Канальцы не справляются с большим количеством белка (им не реабсорбировать такое количество белка, но они пытаются), что выражается в развитии дистрофических изменений в эпителии канальцев. Так или иначе, организм теряет белок, что для развивающегося детского организма является чрезвычайно неблагоприятным фактором. Физическое развитие будет страдать, ибо не из чего делать надстройки растущему организму. Иммунитет, в частности функция антителообразования, будет страдать. Это выражается в частом возникновении и тяжёлом течении бактериальных инфекций у детей.
Фармакокинетика лекарственных препаратов будет несколько иной, чем у других детей, т.к. свободная фракция препарата (не связанного с белком) будет всегда повышенной. Особенно это опасно для часто применяющихся у детей обезболивающих и жаропонижающих препаратов по поводу и без (речь об НПВС,
всеми любимом ибупрофене, парацетамоле, анальгине и запрещённом нынче у детей аспирине и нимесулиде). Одним словом, гипопротеинемия у детей создаёт массу проблем. А если это ещё нефротический синдром в рамках синдрома Пирсона, так вообще туши свет.
Синдром является достаточно серьёзным, т.к. развитие нефротического синдрома происходит в первые месяцы жизни. Заболевание характеризуется высокой летальностью, что становится вполне понятным, если знать, что у таких детей развивается морфологическая картина, очень похожа на мезангиально – пролиферативный гломерулонефрит (быстро прогрессирубщий гломерулонефрит с характерными «полулуниями» в толще гломерулярной капсулы, как правило, не
оставляющего шансов). 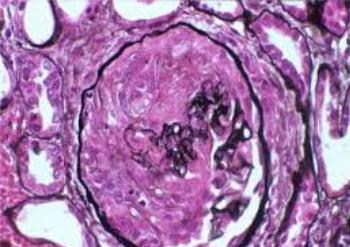
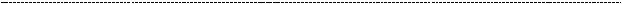
283
(на снимке видно, что клубочек попросту зарастает мезангием. Сосуды от такой наглости редуцируются, в таких условиях адекватная работа почки невозможна).
На фоне быстро прогрессирующей почечной недостаточности в грудном возрасте,
внепочечные проявления мало кого заботят. Но вот что пишут исследователи,
проводившие аутопсию детей, погибших от синдрома Пирсона, про изменения органа зрения: «узкие, не реагирующие на свет и атропин, зрачки вследствие аплазии или атрофии m. dilatator pupillae. Аутопсия, однако, продемонстрировала,
что глазной фенотип не ограничивается только микрокорией и представлен комплексом пороков развития, включая аномалии хрусталика, атрофию цилиарных мышц, изменения роговицы и сетчатки» * (см. библиографический список, 28).
Синдром Пирсона является однйо из частых причин врождённого нефротического синдрома, наряду с нефротическим синдромом финского типа (нефрин – дефицитный), стероид – резистентного подоцин – дефицитного вариантами, но их описание выходит за пределы данной главы и моей компетенции.
16.4.3. Ламинин и мерозин – дефицитная врождённая мышечная дистрофия
Ламинины находят своё пристанище во всех базальных мембранах организма.
Отдельные виды ламинина, как уже было сказано, способны занимать свою нишу,
что характерноидляламинина–211(2а,1ви1–гаммацепь),кодируемый LAMA2, LAMB1 и LAMC1 – генами. Ламинин – 211 – он же мерозин - основная изоформа ламинина в базальной мембране скелетных мышц человека. Ламинин-211
связывается с гликозилированным α-дистрогликаном (DAG1), интегрином α7/β1 и
с другими рецепторами поверхности мышечных клеток, чем обеспечивает структурную организацию мышечной ткани (ну или вносит свой вклад в неё).
Отсутствие мерозина обусловливает тяжелейшую мышечную слабость с самого рождения, с первых недель жизни, которая проявляется мышечной гипотонией,
ограничением движений, замедлением моторного развития ребёнка. И если ребёнок выжил на первом году, далее его поджидает опасность респираторных инфекций из – за слабости дыхательных мышц и нарушения внешнего дыхания.

284
Страдает мимическая мускулатура (слабое сосание), жевательная мускулатура, а
также гладкомышечная ткань кишечника, что создаёт комплексное нарушение питания и усугубляет и без того тяжёлое физическое развитие ребёнка. Но и на этом беды не кончаются.
Но бывает и частичный дефицит мерозина. В этом случае все описаныне выше нарушения более сглаженны и ребёнок имеет больший шанс на выживание.
Как видно на изъятом из https://www.researchgate.net/figure/Schematic-of-the-cardiac-cytoskeletal-
network-The-427-kDa-protein-dystrophin-links_fig5_23385268 художественном шедевре, в
структурной организации мышечной ткани участвует большое количество белков,
и мутации в генах ламинина – 211 – лишь один из возможных вариантов среди
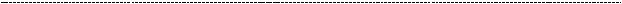
285
причин врождённой мышечной дистрофии. Она может быть обусловлена и дефектом дистрофина (дистрофия Дюшена – Беккера), и в коллагене VI типа
(болезнь Ульриха), и дефицитом интегрина – а7/в1, к которому крепится ламинин.
Вариантов достаточно для изучения.
Болезнь Ульриха является одним из множества вариантов коллагенопатий,
которых мы коснулись в параграфе 16.2. Моё внимание данное заболевание привлекло тем, что оно является одной из причин врождённого вывиха бедра,
врождённой косолапости. Эти заболевания встречаются часто, но их причина зачастую остаётся не выясненной. Такие дети нуждаются в ортопедическом и физиотерапевтическом лечении с самых первых дней. Примечательно, что коллагенопатия VI клинически часто сочетается с гипермобильностью суставов,
и, в отличии от мерозин – негативной дистрофии, не характеризуется значимым ростом КФК – ММ, которая, как ты помнишь из главы 10, является чувствительным маркером поражения мышечной ткани. В остальном, болезьн Ульриха чревато теми же изменениями и нарушениями, что и патология ламинина – 211. Такие дети живут недолго и несчастливо. Хотя кто я такой,
чтобы говорить за них. Только вот мне не весело от одной мысли об их страданиях.
p.s. под пунктом 29 библиографического списка в конце книги указан источник, на который я опирался при написании данного параграфа. Там можно больше прочитать про мышечные дистрофии, та статья просто огонь, автору счастья и здоровья
286
16.5. Самые яркие эпизоды из жизни интегринов
16.5.1. Тромбоцитопатия Гланцмана
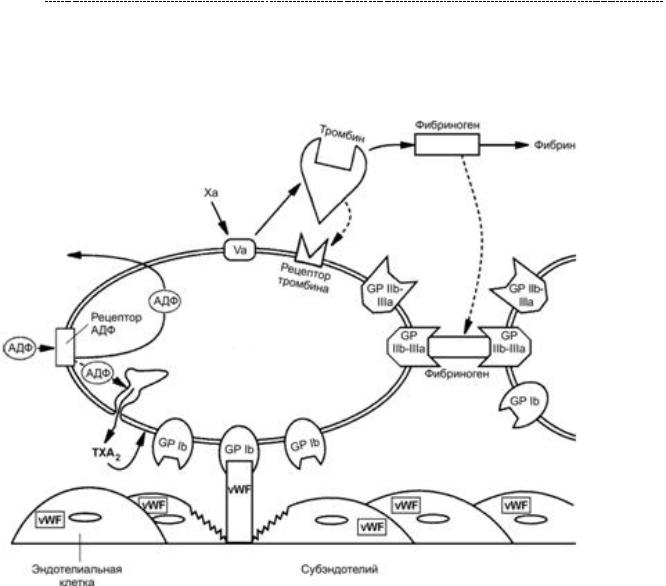
На данной картинке изображена скромная схема тромбоцитарного гемостаза. Нас интересуют сейчас GP – IIb/IIIa – интегриновые рецепторы, которые способны связывать фибриноген. Связывание фибриногена обеспечивает агрегацию тромбоцитов в единый тромбоцитарный сгусток, что является неотъемлимым элементом тромбоцитарного гемостаза. Без такого связывания, тромбоцитарный сгусток расклеится к чёртовой матери или вовсе не будет образовываться.
Соответственно, можно умереть от кровотечения, что иногда и случается.
Такиепациентыимеюточеньнасыщеннуюжизнь.Унихзапростоможетслучиться спонтанное носовое и маточное (не важно, пацан ты или нет) кровотечение.
Описывают и эпизоды желудочно – кишечного кровотечения, что само по себе внушает страх, независимо от причины. Такихпациентов спокойно и спонтанно не прооперируешь, да им даже зубы выбивать нельзя, опасно развитием
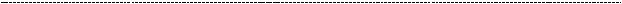
287
кровотечения. Малейшее механическое воздействие на кожу неизбежно приведёт к образованию петехий. Если хочешь блеснуть на этой теме умными словами,
запомни «петехиальный» или микроциркуляторный тип кровоточивости: именно он характерен для патологии тромбоцитарного звена гемостаза, к которому относится и настоящее заболевание.
16.5.2. Интегрины и канцерогенез. Связь с гипертиреозом.
Впоследнее десятилетие случилось достаточно интересное открытие:
продемонстирована связь гипертиреоза с повышенным риском возникновения карциномы самой различной локализации, в частности яичников, молочной и толстой кишки. И некоторое время не могли понять, почему. А потом кааак поняли…
Ты наверняка что – то слышал про гормоны щитовидной железы, про их неотъемлимую роль в биоэнергетике клетки: они усиливают экспрессию генов,
кодирующих натрий – калиевой АТФ – азу, ряд ферментов окисления углеводов и липидов, тем самым увеличивая расход АТФ и интенсифицируя процесс получения энергии, в том числе с помощью ЦТК. Конечно, побочным эффектом такого влияния является повышение потребления кислорода (во многом из – за того, что тиреоидные гормоны являются разобщителями окислительного фосфорилирования и работы ЭТЦ), что не всегда хорошо (привет, ИБС), ну да ладно. Так или иначе, главную роль приписывают именно их влиянию на биоэнергетику клеток.
Но оказалось, что помимо сугубо геномного механизма действия и влияния на биоэнергетику, (главным образом, опосредованно Т3 - трийодтиронином)
существуют ещё и негеномные эффекты, опосредованные, главным образом, Т4 (тироксином или тетрайодтиронином). Вернёмся к уже знакомой схеме.
288
Напомню, что сохранение жизнеспособности клеток многих типов нуждается в интегрин-опосредованной адгезии к внеклеточному матриксу (о чём мы говорили в парагрфае 16.1.) иначе они подвергаются апоптозу. Известно, что интегрины контролируют фиксацию и адгезию клеток, но, как мы уже обсуждали, есть и сигнальная функция интегринов. Для переживания клеток, высокочувствительных к апоптозу, может быть необходимым захват как рецепторов для интегринов, так и рецепторов для факторов роста. Интегрины различны по способности усиливать жизнеспособность клетки. И в этом отношении антиапоптическое действие интегринаCD51/CD61(αVβ3)болеевыражено. Значениеэтогофеноменавтом,что гибель клеток, лишенных надлежащих адгезивных контактов, создает условия для нормального морфогенеза. И тут начинается самое интересное.
Оказалось, что тироксин (Т4) является лигандом для интегрина avB3 (CD51/61).

289
И в условиях высокой концентрации тироксина, его взаимодействие с данным лигандом будет столь же высоким. Как мы видим, это приведёт к активации SRC –
киназы,что чреватоактивациейМАРК– митогенногокаскада,т.е.кпролиферации клетки. Есть данные, что и трийодтиронин способен оказывать антиапоптотическое действие путём прямого активирование Src и PI3K – киназ. * (пункт 30 в библиографическом списке). Как бы там ни было, результат один:
тиреоидные гормоны оказывают антиапоптотическое дейсвтие, что лежит в основе их проканцерогенного эффекта при гипертиреозе.
16.6. Мукополисахаридозы.
Мы завершаем своё путешествие по закаулкам соединительной ткани, и у нас остался финальный босс – мукополисахаридозы. Заболевание на слуху далеко не у каждого студента, в силу его нечастой встречаемости, но колоссальнейшего значения. Мукополисахаридоз, как ФКУ или врождённый гипотиреоз, нужно начинать лечить как можно раньше, так как накону адекватое развитие ребёнка.
Только и тут не обошлось без сложностей, которых мы далее коснёмся.
Мукополисахаридозы (МПС) являются одним из вариантов болезней накопления и связаны с дефектом (качественным или количественным) лизосомальных гликозидаз, расщепляющих углеводные связи в углеводном компоненте протеогликанов, т.е. занимающихся катаболизмом гликозаминогликанов.
На сегодняшний день известно свыше 15 типов МПС, каждый из которых обусловлен дефектом конкретного фермента, конкретной гликозидазы. Я приведу лишь несколько из них. Все они характеризуются сходными клиническими проявлениями, отличительной чертой которых является мультисистемность поражений. Практически ни один орган не остаётся незатронутым, но обо всём по порядку.
290
Тип |
Название |
Фермент |
Ген |
Тип |
Лечение |
наследования |
|||||
|
Синдромы |
|
|
|
|
МПС I |
Гурлера, Шейе |
α-L-идуронидаза |
IDUA |
Аутосомно- |
Ларонидаза |
или Гурлера- |
4p16.3 |
рецессивный |
|||
|
Шейе |
|
|
|
|
|
|
|
|
|
|
МПС II |
Синдром |
Идуронат-2-сульфатаза |
IDS |
X-сцепленный |
Идурсульфаза |
Хантера |
Xq28 |
рецессивный |
|||
|
|
|
|
|
|
МПС IIIA |
Синдром |
Гепаран-N-сульфатаза |
SGSH |
Аутосомно- |
Разрабатывается |
Санфилиппо A |
17q25.3 |
рецессивный |
|||
|
|
|
|
|
|
МПС IIIB |
Синдром |
α-N- |
NAGLU |
Аутосомно- |
|
Санфилиппо В |
ацетилглюкозаминидаза |
17q21 |
рецессивный |
|
|
|
|
|
|
|
|
|
Синдром |
Ацетил-КоА α- |
HGSNAT Аутосомно- |
|
|
МПС IIIC |
глюкозамин-ацетил- |
|
|||
Санфилиппо С |
8p11.1 |
рецессивный |
|
||
|
|
трансфераза |
|
|
|
МПС IIID |
Синдром |
N-ацетилглюкозамин-6- |
GNS |
Аутосомно- |
- |
Санфилиппо D |
сульфатаза |
12q14 |
рецессивный |
||
|
|
|
|
|
|
MПС IVA |
Синдром |
Галактозамин-6 |
GALNS |
Аутосомно- |
Элосульфаза |
Моркио А |
сульфатсульфатаза |
16q24.3 |
рецессивный |
||
|
|
|
|
|
|
MПС IVB |
Синдром |
β-Галактозидаза |
GLB1 |
Аутосомно- |
- |
Моркио В |
3p21.33 |
рецессивный |
|||
|
|
|
|
|
|
MПС VI |
Синдром |
Арилсульфатаза B |
ARSB |
Аутосомно- |
Галсульфаза |
Марото-Лами |
5q11.q13 |
рецессивный |
|||
|
|
|
|
|
|
МПС VII |
Синдром Слая |
β-Глюкуронидаза |
GUSB |
Аутосомно- |
Разрабатыва |
7q21.11 |
рецессивный |
||||
|
|
|
|
|
|
|
Синдром |
|
AH |
Аутосомно- |
|
МПС IX |
Гиалуронидаза I |
3p21.3- |
- |
||
|
Натовича |
|
p21.2 |
рецессивный |
|
|
|
|
|
|
|
На данной таблице представлены названия каждого варианта МПС, затронутый фермент, тип наследования и наличие или отсутсвтие заместительной энзимотерапии.
Патофизиологические аспекты (куда же без них).
ГАГ, как я уже упоминал, очень гидрофобны и притягивают на себя воду в силу своего строения и наличия очень большого количества полярный ОН – групп. Это лежитвосновенарушенияобразованиясоединительнойткани,втом числесинтеза коллагена: его откладывается тоже очень много.