

2. Поликонденсация в растворе. Роль растворителя в процессе
Использование растворителя резко снижает вязкость реакционной системы и исключает (или существенно снижает) диффузионные ограничения во взаимодействии функциональных групп на завершающих стадиях процесса. Это приводит к повышению как скорости процесса, так и глубины превращения и способствует образованию более высокомолекулярных продуктов. Кроме того, по сравнению с поликонденсацией в расплаве улучшается теплообмен, исключаются местные перегревы реакционной смеси и связанные с ним деструктивные процессы. Недостатки при поликонденсации в растворе:
Побочные реакции с участием растворителя;
Меньшая эффективность использования объема реакционной аппаратуры;
Наличие дополнительных стадий осаждения полимера, регенерации и очистки растворителя.
Ароматические полиамиды и ароматические полиэфиры, полиимиды могут быть получены и переработаны только при поликонденсации в растворе. Например, поли-п-фенилентерефталамид получают поликонденсацией п-фенилендиамина и дихлорангидрида терефталевой кислоты в среде N,N`-диметилацетаамида с добавлением неорганических солей:

Растворитель при поликонденсации в растворе растворяет исходные мономеры и образующиеся полимеры, облегчает удаление побочного НМС (отгонка в виде азеотропа, химическое связывание), ускоряет основную реакцию ступенчатого роста цепей, изменяет в соотношении реакции роста цепи и циклизации.
При поликонденсации с выделением воды в качестве НМС часто используют растворители, образующие с ней низкокипящие азеотропные смеси (например добавляют толуол, вода отгоняется при Т=84,1 °С). Вода удаляется быстро и более полно, а поликонденсационное равновесие смещается в сторону образования ВМС. Такой тип поликонденсации называется азеотропной поликонденсацией.
При выделении HCl в качестве НМС, ее связывают амидными растворителями (диметилформамид, диметилацетамид, N-метилпирролидон), а также третичными аминами (пиридин, триэтиламин). В их отсутствии HCl вступает в реакцию с исходными диаминами с образованием нереакционноспособных или малоактивных дигидрохлоридов.
Природа растворителя сложным образом связана со скоростью основной реакции роста цепи при поликонденсации: обычно скорость этого процесса определяется полярностью растворителя, его сольватирующей способностью и термодинамическим качеством по отношению к образующемуся полимеру. В зависимости от природы растворителя скорость реакции может изменяться на три порядка. Аналогичные эффекты проявляются и при конденсации мономеров; они способствуют повышению глубины превращения и образованию более высокомолекулярных полимеров.
Но также возможно понижение константы скорости вследствие повышения вязкости раствора, так и в случае, когда используемый растворитель является плохим – макромолекулы сворачиваются в клубки, внутри которых могут оказаться концевые функциональные группы, не способные к последующим реакциям роста цепей из-за диффузионных ограничений. На рисунке приведена зависимость константы скорости синтеза поликарбоната поликонденсацией дифенилолпропана с его бисхлорформиатом от диэлектрической проницаемости ε смешанного растворителя:


Прямые 1 и 2 отличаются лишь природой второго компонента в смеси растворителей: дихлорбензол понижает только диэлектрическую проницаемость среды и не влияет на конформационное строение молекулы в растворе; декалин также понижает диэлектрическую проницаемость, но одновременно, являясь осадителем для полимера, способствует сворачиванию его макромолекул в клубки, плотность которых возрастает с увеличением доли декалина в смеси. Вместе с увеличением доли декалина (уменьшением диэлектрической проницаемости среды) резко уменьшается скорость поликонденсации – при таком же значении ε в смеси с n-дихлорбензолом она на 1,5 порядка выше. В тоже время скорость реакции полимеробразования остается ниже скорости модельных реакций НМС, причем на последнюю не влияет природа смеси растворителей. С понижением концентрации мономера в растворе возрастает доля реакций циклизации из-за уменьшения вероятности межмолекулярных взаимодействий функциональных групп. Однако в зависимости от природы растворителя доля реакций циклизации может существенно изменяться, что видно на примере выхода циклического лактама в процессе поликонденсации в среде различных растворителей 4-тио-ε-аминокапроновой кислоты:

Также растворитель может изменять константу поликонденсационного равновесия, а также интенсивность побочных реакций мономеров.
3 ПАП – хим. р-ция на полимере, протекающая без изменения его ММ. В общем виде его можно представить схемой:
в кот. А и Б – исх. и конечная ФГ; R – реагент; Х – побочное НМС; Е – группа, образовавшаяся вследствие побочного превращения.
Поскольку реакц. группы явл. частью макромол-лы, нельзя отделить целевые продукты от исх. в-ва, часто не удается достигнуть полного превращения, а побочные р-ции протекают на той же макромол-ле.
Неполное превращение в макромол-ле м.б. следствием стерических затруднений, а иногда (когда в р-ции участвуют 2 соседние группы) обусловливается и статическими причинами – образованием изолированных ФГ, например при ацеталировании ПВС:
или при отщеплении хлора от ПВХ в присутствии цинка:
Для таких р-ций по расчету Флори максимально возможное статистическое превращение составляет 86,5%. Однако отщепление мол-л НМС от макромол-л может протекать с изменением природы основной цепи:
Для многих ПАП принцип равной реакционной спос-сти ФГ не применим по след. причинам:
доступность ФГ;
влияние соседних групп;
конфигурационный и конформационный эффекты;
электростатический эффект;
морфологический эффект;
кооперативный эффект;
эффект негомогенной активности;
влияние длины цепи и конц-ции.
В чистом виде каждый из них проявляется редко; обычно реакц. спос-сть ФГ опред-ся суммарным д-ем нескольких из них.
1. Доступность ФГ обусловлена в основном стерическими препятствиями, кот. существенно выше при наличии объёмистых групп-к в полимерной цепи по сравнению с теми же групп-ками в НМС.
Ограниченная доступность ФГ м.б. обусловлена и конформационным эффектом, например повышение жесткости цепи в п-се циклизации полиамидокислоты в полиамиды приводит к уменьш-ию скорости р-ции с конверсией.
2.Влияние соседних групп – наиболее частый эффект, кот. замедляет или ускоряет р-ции ФГ, соеди-ненных с основной цепью макромол-л. При исследовании щелочного гидролиза ПВА установлено повышение скорости в ряду триад:
Ацетатная группа, располож. между двумя ОН-группами, омыляется в 100 раз быстрее, чем находящаяся между двумя ацетатными. Причина этого – адсорбция каталитически активных ионов ОН- на образующихся гидроксильных группах, что повышает локальную конц-цию щелочи в районе омыляемой группы и ускоряет р-цию.
3.Конфигурационный и конформационный эффекты. Характер соединения звеньев в цепи и их пространственное расположение сущ-но влияют на скорость ПАП. Например, изотактические п-ры реагируют быстрее синдиотактических или атактических.
У гибкоцепных макромол-л доступность для НМС их ФГ будет зависеть от характера взаимод-ия п-ра с р-лем. В хороших р-лях разбухание клубков улучшает их проницаемость для НМС и повышает скорость р-ции. В этом случае скорость р-ции опред-ся не хим. активностью ФГ, а диффузионными ограничениями, обусловленными конформационным состоянием ВМС. На практике влияние конформации прослеживается путем осуществления одного и того же ПАП в р-лях, отлич. по т/д качеству в отношении полимерного субстрата.
4. Электростатический эффект: взаимод-ие одинаково или различно заряженных групп в цепи может как замедлять, так и ускорять ПАП. В р-ции щелочного гидролиза полиакриламида электростати-ческое отталкивание отрицательно заряженными карбоксилатными группами гидроксид-иона способствует замедлению процесса(совместное проявление с эффектом соседней группы). Иногда одинаково заряженные группы, отталкиваясь, разворачивают макромолекулярный клубок, что облегчает взаимод-ие непрореагировавших групп с реагентом (совместное проявление с конформац. эффектом).
5.Кооперативный эффект обусловлен вторичными взаимод-ями между участками макромол-л субстрата и фрагментами мол-л реагента, не участвующими в основном хим. взаимод-ии. Схематически это выглядит так:
ФГ А полимерной цепи явл. реакционным центром и взаимод-ет с ФГ М реагента. При этом взаимод-ют между собой группа А п-ра и вторая ФГ N реагента (кулоновское взаимод-ие, водородная связь). Вторичное взаимод-ие А…N стабилизирует переходное состояние, образованное группами А и М, и ускоряет тем самым основную р-цию.
6.Морфологический эффект проявляется только при гетерогенных ПАП. Аморфный полимер, сильно набухая в реакц. среде, быстрее подвергается ПАП, чем кристаллический. Ассоциация макромол-л в р-ре уменьшает доступность ФГ и снижает скорость р-ции.
7. Эффект негомогенной активности проявляется в побочном связывании НМ реагента полимерной матрицей и «исключением» его из основной р-ции. Здесь имеется в виду связывание реагента за счет Н-связей, ван-дер-ваальсовых сил; реагент оказывается «иммобилизованным» около опреде-ленных участков макромол-л и не вступает в основное взаимод-ие по ФГруппам.
8. Влияние длины цепи и конц-ции: с повышением ММ п-ра размеры клубков и вязкость р-ра возрастают, что увеличивает диффузионные ограничения и неравномерности в распределении конц-ции реагирующих групп (конц-ция НМ реагента внутри клубков всегда меньше средней по реакц. объёму).
Билет 9
Полимеризация - цепной процесс, включающий в себя три стадии: инициирование (образование активных центров); рост цепи вследствие присоединения молекул мономера к образовавшимся активным центрам; обрыв цепи в результате реакции активного центра с инициатором, другими активными центрами, с примесями или со специально вводимыми веществами.
Общая схема полимеризации не зависимо от природы активного центра может быть представлена следующим образом (* - активный центр):
I I*, I* + M IM* (инициирование)
IM* + М IMM*…I(M)nM* (рост цепи)
I(M)nM* +*M(M)mI I(M)nMM(M)mI (рекомбинация)
I(M)nM* + M I(M)n+1 + M* (передача цепи)
I(M)nM* неактивный полимер (обрыв цепи).
Центрами роста могут быть свободные радикалы, анионы, катионы, комплексные соединения.
Необходимые для начала радикальной полимеризации инициирующие свободные радикалы получают:
введением в мономер веществ, способных распадаться с образованием радикалов;
генерацией свободных радикалов нагреванием мономера; облучением мономера УФ-светом; воздействием на мономер γ- и рентгеновских лучей, ускоренных электронов.
Инициаторы свобонорадикальной полимеризации – вещества, распадающиеся при умеренных температурах с образованием свободных радикалов.
Название |
Схема распада |
Температурный интервал, °С |
Распад по связи О-О |
||
Гидроперекись третичного бутана |
(CH3)3COOH (CH3)3CO* +*OH *CH3 + (CH3)2-C=O |
80-140 |
Гидроперекись кумола |
C6H5(CH3)2COOH C6H5* + (CH3)2C=O |
80-140 |
Перекись бензоила |
(C6H5-COO)2 2C6H5 COO* 2C6H5* + 2CO2 |
100-170 |
Перекись водорода |
H2O2 2HO* |
40-80 |
Персульфаты щелочных металлов, NH4 |
Na2S2O8 2*SO4 (ион-радикал) |
50-70 |
Пероксидкарбонаты R=C6H11 |
ROC(O)-O-O-C(O)OR 2RO + 2CO2 |
10-80 |
Распад по связи С-N |
||
Динитрил азодиизомасляной кислоты |
NC-C(CH3)2-N=N-C(CH3)2-CN 2NC-C+(CH3)2 + N2 |
50-80 |
Распад по связи N-N |
||
Диазоаминосоединения |
C6H5NHN=NR C6H5NH* + R* + N2 |
50-120 |
Распад по связи С-С |
||
Гескахлорэтан |
Cl3C-CCl3 2CCl3* |
100 |
Гексафенилэтан |
Ph3C-CPh3 2CPh3* |
0-100 |
Обычно часть радикалов, образовавшихся при распаде инициатора, расходуется в побочных реакциях, не связанных с полимеризацией, образуя неактивные продукты. Например:

Распад инициаторов ускоряется в присутствии специальных добавок – промоторов, вступающих в реакцию с инициатором. (Fe+2, третичные амины, сульфиты). Например:
HOOH + Fe+2Fe+3 + OH* + OH-
ROOR + Fe+2 RO* + RO- + Fe+3
ROOH + Fe+2RO* + OH- + Fe+3
RO* + Fe+2 RO- + Fe+3
S2O8-2 + Fe+2 *SO4- + SO42- + Fe+3
*SO4- + H2O OH* + HSO4-
ЭД заместители, введенные в молекулу перекиси бензоила, увеличивают электронную плотность бензольных колец и снижают устойчивость соединения, повышая скорость инициирования. ЭА заместители (NO2, CN) повышают устойчивость инициатора, снижают скорость инициирования.
Эффективными инициаторами полимеризации являются азосоединения, распадающиеся легче перекисных.
Если вводимое в полимеризующуюся систему вещество АВ реагирует с инициирующими радикалами (R*): R* + AB = RA + B* или с растущими макрорадикалами ~R* с образованием радикалов В*, не способных инициировать полимеризацию исходного мономера, то вещество АВ является ингибитором. При этом радикалы В* могут взаимодействовать с R* и ~R* с обрывом цепи. Например, полимеризация стирола ингибируется гидрохиноном. Исходный ингибитор – бензохинон (продукт окисления гидрохинона). Бензохинон реагирует с инициирующими и растущими радикалами с образованием радикала семихинона:
Из-за сопряжения неспаренного электрона с π-электронами бензольного кольца радикал семихинона малоактивен и не инициирует полимеризацию стирола, хотя и реагирует с растущим полимерным радикалом, обрывая цепь:
Наибольшим ингибирующим эффектом обладают соединения, реагирующие с инициирующим радикалом (свободные радикалы, неактивные для инициирования полимеризации, но реагирующие с инициирующими радикалами), например, трифенильный радикал Ph3C.
Наряду с реакциями функциональных групп мономеров и олигомеров, приводящими к росту цепи в поликонденсационном процессе, возможны и другие превращения функциональных групп, чаще всего – реакции циклизации и обмена.
Реакции циклизации:

Например, при поликонденсации оксикислот HO(CH2)xCOOH с x=3 и x=4 преимущественно образуются циклические лактоны:

Мономеры типа а-а и b-b – этиленгликоль и диэтилоксалат – при взаимодействии также образуют циклический продукт:

При поликонденсации гексаметилендиамина и адипиновой кислоты наряду с линейным полимером образуются макроциклы с m=20-30:

При синтезе полисульфонов по схеме:

Образуется частично димер:

Возможность образования циклов при поликонденсации определяется совокупностью термодинамических и кинетических факторов, основные из которых:
Угловое напряжение образующегося цикла при наличии в нем напряженных углов между связями.
Напряжение внутреннего заполнения (трансаннулярное), возникающее в результате взаимодействия замещенных атомов или радикалов, расположенных внутри цикла и на его противоположных сторонах.
Стерические препятствия из-за взаимодействия боковых заместителей цикла.
Совместное действие этих факторов на примере циклов, состоящих из CH2-групп, иллюстрирует рис.:

На участке АБ способность к циклизации возрастает из-за уменьшения углового напряжения, на участке БВ она уменьшается в связи с действием внутреннего заполнения, на участке ВГ снова увеличивается из-за постепенного исчезновения пространственных затруднений. Для углеродных циклов с другими, нежели атомы H, заместителями и для гетероциклов зависимость аналогична, но точки А,Б,В,Г будут отвечать циклам иного размера.
Из кинетических факторов, влияющих на циклизацию, следует отметить, концентрацию в температуру. С повышением концентрации мономеров в системе усиливаются межмолекулярные взаимодействия и вероятность циклизации уменьшается. Энергия активизации циклизации обычно выше энергии активации роста цепи, поэтому повышение температуры при прочих равных условиях будет способствовать циклизации.
Обменные реакции. В ходе обратимой поликонденсации происходит не только взаимодействие функциональных групп друг с другом, но и реакции этих групп с уже образовавшимися межзвенными связями олигомерных и полимерных молекул. На начальных стадиях поликонденсации, когда содержание мономеров в реакционной смеси еще велико, возможны следующие превращения:
а) при синтезе полиэфиров из дикарбоновых кислот и гликолей вследствие взаимодействия карбоксильной группы дикарбоновой кислоты со сложноэфирной связью (реакции ацидолиза) цепи разрываются и образуются два и более коротких фрагмента:
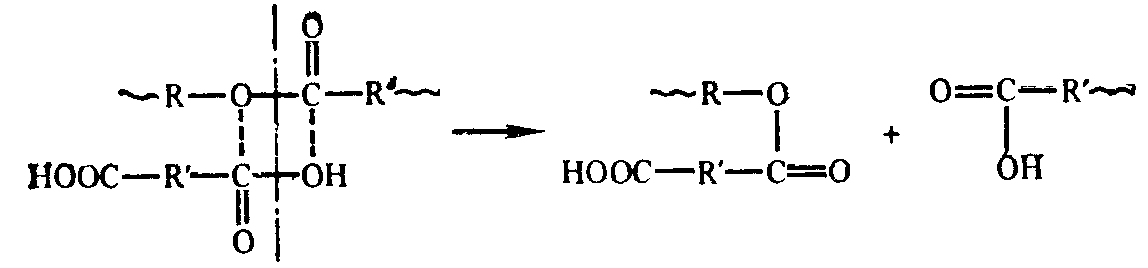
Аналогичная реакция может произойти и под действием гликоля (алкоголиз):

б) при образовании полиамидов наряду с ацидолизом образующихся цепей по амидным связям возможно их расщепление под действием аминогрупп мономера (аминолиз):

В случае обменных реакций с участием функциональных групп мономеров происходит уменьшение молекулярной массы образующегося полимера. По мере исчерпания мономеров в обменные реакции ацидолиза, алкоголиза или аминолиза вступают концевые функциональные группы образующихся олигомеров и полимеров:
Ацидолиз полиамида:

Алкоголиз полиэфира:

Аминолиз полиамида:

Характерной особенностью обменных реакций с участием концевых групп олигомеров и полимеров является то, что число цепей от такого взаимодействия не меняется (из двух образуется двое), но меняется молекулярно-массовое распределение. Вероятность участия в них более длинных цепей выше (у них большее число межзвенных связей – сложноэфирных, амидных). Поэтому оцидолиз, аминолиз и алкоголиз выравнивает длину цепей, приближая молекулярно-массовое распределение к наиболее вероятному.
Макромолекулы могут реагировать друг с другом и без участия концевых функциональных групп непосредственно межзвенными связями (реакции межцепного обмена). Например, при взаимодействии макромолекул сложного полиэфира происходит переэтерификация:

А при взаимодействии макромолекул полиамида – переамидирование:

При использовании двух различных по природе полиэфиров или полиамидов на начальной стадии межцепного обмена возможно образование блок-сополимеров; при большей дилтельности этой реакции могут быть получены статические сополимеры. В случае сополимеров в результате межцепного обмена выравнивается молекулярно-массовое распределение.
Использование растворителя резко снижает вязкость реакционной системы и исключает (или существенно снижает) диффузионные ограничения во взаимодействии функциональных групп на завершающих стадиях процесса. Это приводит к повышению как скорости процесса, так и глубины превращения и способствует образованию более высокомолекулярных продуктов. Кроме того, по сравнению с поликонденсацией в расплаве улучшается теплообмен, исключаются местные перегревы реакционной смеси и связанные с ним деструктивные процессы. Недостатки при поликонденсации в растворе:
Побочные реакции с участием растворителя;
Меньшая эффективность использования объема реакционной аппаратуры;
Наличие дополнительных стадий осаждения полимера, регенерации и очистки растворителя.
Ароматические полиамиды и ароматические полиэфиры, полиимиды могут быть получены и переработаны только при поликонденсации в растворе. Например, поли-п-фенилентерефталамид получают поликонденсацией п-фенилендиамина и дихлорангидрида терефталевой кислоты в среде N,N`-диметилацетаамида с добавлением неорганических солей:
Растворитель при поликонденсации в растворе растворяет исходные мономеры и образующиеся полимеры, облегчает удаление побочного НМС (отгонка в виде азеотропа, химическое связывание), ускоряет основную реакцию ступенчатого роста цепей, изменяет в соотношении реакции роста цепи и циклизации.
При поликонденсации с выделением воды в качестве НМС часто используют растворители, образующие с ней низкокипящие азеотропные смеси (например добавляют толуол, вода отгоняется при Т=84,1 °С). Вода удаляется быстро и более полно, а поликонденсационное равновесие смещается в сторону образования ВМС. Такой тип поликонденсации называется азеотропной поликонденсацией.
При выделении HCl в качестве НМС, ее связывают амидными растворителями (диметилформамид, диметилацетамид, N-метилпирролидон), а также третичными аминами (пиридин, триэтиламин). В их отсутствии HCl вступает в реакцию с исходными диаминами с образованием нереакционноспособных или малоактивных дигидрохлоридов.
В зависимости от природы растворителя скорость реакции может изменяться на три порядка. Аналогичные эффекты проявляются и при конденсации мономеров; они способствуют повышению глубины превращения и образованию более высокомолекулярных полимеров.
Но также возможно понижение константы скорости вследствие повышения вязкости раствора, так и в случае, когда используемый растворитель является плохим – макромолекулы сворачиваются в клубки, внутри которых могут оказаться концевые функциональные группы, не способные к последующим реакциям роста цепей из-за диффузионных ограничений.
Также растворитель может изменять константу поликонденсационного равновесия, а также интенсивность побочных реакций мономеров.
В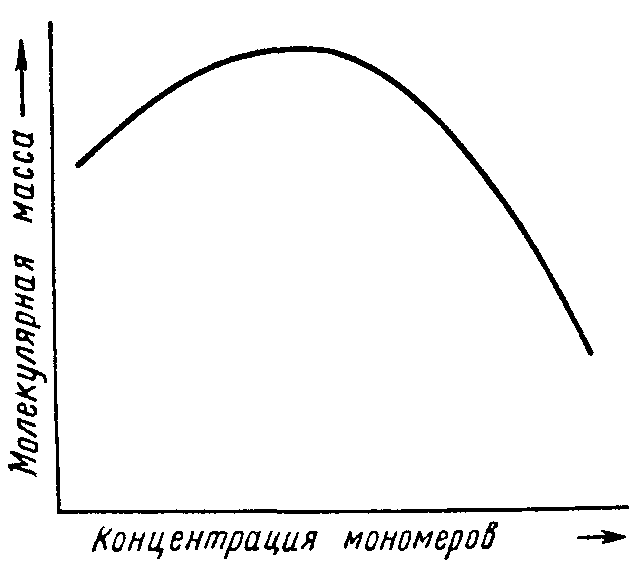 лияние
концентрации мономеров. Обычно
максимальная молекулярная масса полимера
достигается при средних значениях
концентрации реагирующих веществ:
лияние
концентрации мономеров. Обычно
максимальная молекулярная масса полимера
достигается при средних значениях
концентрации реагирующих веществ:
При низких концентрациях мономеров уменьшение молекулярной массы связано с увеличением относительной доли примесей и монофункциональных соединений; при высоких концентрациях рост цепей прекращается вследствие возрастания вязкости и связанных с ней диффузионных ограничений. Максимальная молекулярная масса при поликонденсации в растворе достигается при эквимольном соотношении функциональных групп реагирующих мономеров.
Билет 10
1. Обменные реакции. В ходе обратимой поликонденсации происходит не только взаимодействие функциональных групп друг с другом, но и реакции этих групп с уже образовавшимися межзвенными связями олигомерных и полимерных молекул. На начальных стадиях поликонденсации, когда содержание мономеров в реакционной смеси еще велико, возможны следующие превращения:
а) при синтезе полиэфиров из дикарбоновых кислот и гликолей вследствие взаимодействия карбоксильной группы дикарбоновой кислоты со сложноэфирной связью (реакции ацидолиза) цепи разрываются и образуются два и более коротких фрагмента:
Аналогичная реакция может произойти и под действием гликоля (алкоголиз):
б) при образовании полиамидов наряду с ацидолизом образующихся цепей по амидным связям возможно их расщепление под действием аминогрупп мономера (аминолиз):
В случае обменных реакций с участием функциональных групп мономеров происходит уменьшение молекулярной массы образующегося полимера. По мере исчерпания мономеров в обменные реакции ацидолиза, алкоголиза или аминолиза вступают концевые функциональные группы образующихся олигомеров и полимеров:
Ацидолиз полиамида:
Алкоголиз полиэфира:
Аминолиз полиамида:
Характерной особенностью обменных реакций с участием концевых групп олигомеров и полимеров является то, что число цепей от такого взаимодействия не меняется (из двух образуется двое), но меняется молекулярно-массовое распределение. Вероятность участия в них более длинных цепей выше (у них большее число межзвенных связей – сложноэфирных, амидных). Поэтому оцидолиз, аминолиз и алкоголиз выравнивает длину цепей, приближая молекулярно-массовое распределение к наиболее вероятному.
Макромолекулы могут реагировать друг с другом и без участия концевых функциональных групп непосредственно межзвенными связями (реакции межцепного обмена). Например, при взаимодействии макромолекул сложного полиэфира происходит переэтерификация:
А при взаимодействии макромолекул полиамида – переамидирование:
При использовании двух различных по природе полиэфиров или полиамидов на начальной стадии межцепного обмена возможно образование блок-сополимеров; при большей дилтельности этой реакции могут быть получены статические сополимеры. В случае сополимеров в результате межцепного обмена выравнивается молекулярно-массовое распределение.
Катионная полимеризация простых виниловых эфиров общей формулой CH2=CH-OR обнаруживает ряд специфических особенностей:
В карбокатионном центре положительный заряд делокализован между атомами С и О:
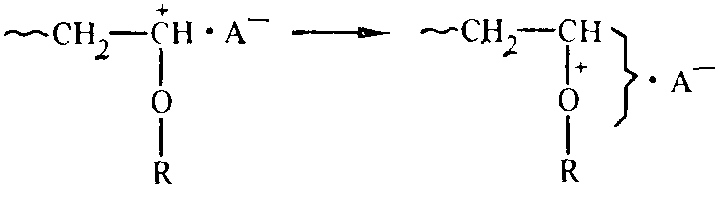
Вследствие сильно выраженного нуклеофильного характера двойной связи простые виниловые эфиры полимеризуются под действием любых катионных катилизаторов, в том числе малоактивного FeCl3.
Процесс проводят в неполярных средах при низких температурах, что обеспечивает высокие молекулярные массы.
Реакции ограничения цепи:
а) передача цепи на мономер:

б) мономолекулярный обрыв с отщеплением активного катионного агента и формирование концевой циклической группировки:

Скорость полимеризации в присутствии комплексов кислот Льюиса равна r=k[M]2[C]. Второй порядок по мономеру обусловлен бимолекулярным обрывом цепи (реакция (а)).
Катионная полимеризация позволяет получать из простых виниловых эфиров стереорегулярные полимеры. В полярной среде рост цепи идет на связанных ионных парах. Ориентация мономера на ионной паре активного центра с плавным переходом ориентированной молекулы в состав растущей цепи – причина стереорегулирования. При повышении полярности среды, ослабляющей связь катионного центра растущей цепи с противоионом, эффект стереорегулирования исчезает.
Полимераналогичное превращение – химическая реакция на полимере, протекающая без изменения его молекулярной массы. В общем виде его можно представить схемой:

В которой А и Б – исходная и конечная функциональные группы; R – реагент; X – побочное НМС; Е – группа, образовавшаяся вследствие побочного превращения.
Поскольку реакционные группы являются частью макромолекулы, нельзя отделить целевые продукты от исходного вещества, часто не удается достигнуть полного превращения, а побочные реакции протекают на той же макромолекуле. Примером полимераналогичных превращений могут служить следующие реакции:


Для многих полимераналогичных превращений принцип равной реакционной способности групп не применим по следующим причинам, которые в чистом виде проявляются редко, обычно реакционная способность функциональных групп определяется суммарным действием нескольких из них:
Доступность функциональных групп обусловлена в основном стерическими препятствиями, которые существенно выше при наличии объемистых группировок в полимерной цепи по сравнению с теми же группировками в НМС.
Ограниченная доступность функциональных групп может быть обусловлена и конформационным эффектом, например повышение жесткости цепи в процессе циклизации полиамидокислоты в полиамиды приводит к уменьшению скорости реакции с конверсией.
Влияние соседних групп – наиболее частый эффект, который замедляет или ускоряет реакции функциональных групп, соединенных с основной цепью макромолекул. При исследовании щелочного гидролиза ПВА установлено повышение скорости в ряду триад:

Ацетатная группа, расположенная между двумя ОН-группами, омыляется в 100 раз быстрее, чем находящаяся между двумя ацетатными.
Причина этого – адсорбция каталитически активных ионов ОН- на образующихся гидроксильных группах, что повышает локальную концентрацию щелочи в районе омыляемой группы и ускоряет реакцию.
Конфигурационный и конформационный эффекты. Характер соединения звеньев в цепи и их пространственное расположение существенно влияют на скорость полимераналогичный превращений. Например, изотактические полимеры реагируют быстрее синдиотактических или атактических.
У гибкоцепных макромолекул доступность для НМС их функциональных групп будет зависеть от характера взаимодействия полимера с растворителем. В хороших растворителях разбухание клубков улучшает их проницаемость для НМС и повышает скорость реакции. В этом случае скорость реакции определяется диффузионными ограничениями, обусловленными конформационным состоянием ВМС.
Электростатический эффект: взаимодействие одинаково или различно заряженных групп в цепи может как замедлять, так и ускорять полимераналогичные превращения. В реакции щелочного гидролиза полиакриламида электростатическое отталкивание отрицательно заряженными карбоксилатными группами гидроксид-иона способствует замедлению процесса. Иногда одинаково заряженные группы отталкиваясь, разворачивают макромолекулярный клубок, что облегчает взаимодействие непрореагировавших групп с реагентом.
Кооперативный эффект обусловлен вторичными взаимодействиями между участками макромолекул субстрата и фрагментами молекул реагента, не участвующими в основном химическом взаимодействии. Схематически это выглядит так:

Функциональная группа А полимерной цепи является реакционным центром и взаимодействует с функциональной группой М реагента. При этом взаимодействуют между собой группы А` полимера и вторая функциональная группа N реагента. Вторичное взаимодействие A`…N стабилизирует переходное состояние, образованное группами А и М, и ускоряет тем самым основную реакцию.
Морфологический эффект проявляется только при гетерогенных полимераналогичных превращениях. Аморфный полимер, сильно набухая в реакционной среде, быстрее подвергается полимераналогичному превращению, чем кристаллический. Ассоциация макромолекул к растворе уменьшает доступность функциональных групп и снижает скорость реакции.
Эффект негомогенной активности проявляется в побочном связывании низкомолекулярного реагента полимерной матрицей и «исключением» его из основной реакции. Здесь имеется в виду связывание реагента за счет Н-связей, ванн-дер-ваальсовых сил; реагент оказывается «иммобилизованным» около определенных участков макромолекулы и не вступает в основное взаимодействие по функциональным группам.
Влияние длины цепи и кон-ции: с повышением ММ пол-ра размеры клубков и вязкость раствора возрастают, что увеличивает диффузионные ограничения и равномерности в распределении концентрации реагирующих групп (концентрация НМС внутри клубков всегда меньше средней по реакционному объему).
Билет 11.
1. По фазовому состоянию реакционных систем, в которых осуществляют полим-цию м-ра по рад-ному мех-му, процессы разделяют на гомогенные( в массе м-ра или в р-ре) и гетерогенные( в суспензии и в эмульсии). При полим-ции а р-ре полимер не выделяется из реакционной системы, процесс на всех стадиях остается гомогенным.
Полим-ция в массе (блоке) – простейший способ полим-ции чистого м-ра, при котором пол-р не загрязняется побочными продуктами. Но и-за сильного повышения вязкости реакционной системы уже на ранних стадиях процесса и его экзотермичности, а также из-за автоускорения (гель-эффект) сильно затруднен отвод теплоты из зоны р-ции. В рез-те могут возникнуть локальные перегревы, приводящие к деструкции пол-ра и ухудшению его св-в. Возрастает также роль передачи цепи на пол-р, что способствует вместе с локальными перегревами формирования более широкого ММР. Поэтому данный способ применяется ограниченно (этилен, стирол, ММА). Полим-цию этилена проводят при р= 100-300 МПа, Т=190-250°С с конверсией 20%. При полим-ции стирола и ММА тепло отводят в две стадии: вначале при относительно низкой тем-ре (80°С) проводят полим-цию до конверсии 20-40%, получая форполимер (р-р пол-ра в м-ре). Вторую стадию для полистирола проводят в колоннах непрерывного действия с постепенным повышением тем-ры сверху вниз и выгрузкой готового пол-ра снизу. Конверсия достигает 98-100%. Частично заполимеризованный ММА заливают в формы и полим-зуют постепенным повышением тем-ры.
Классическая хим. кинетика рассматривает р-ции в идеализированных условиях, не осложненных процессами тепло- и массопередачи, диффузии и др. При рад-ной полим-ции в массе указанными процессами можно пренебречь лишь на начальной стадии р-ции, когда вязкость реакционной массы увеличивается незначительно.
Через какой-то пром-к времени с начала полим-ции число образующихся рад-лов сравнивается с числом исчезающих макрорад-лов и система переходит в стационарное состояние с постоянной конц-ей рад-лов (d[R˙]/dt=0), а также с постоянной ск-тью роста цепи. Может оказаться, что и в стац-ном состоянии ск-ть процесса изм-ся вследствие изм-ния конц-ции м-ра. Условием стац-ности явл-ся постоянство конц-ции макрорад-лов ~R˙: vp/[M]=kp[R˙], или рав-во ск-тей иниц-ния vи обрыва vоб, или с учетом что vи=kи[In] и vo=ko[R˙]2:
![]()
Ск-ть
полим-ции в стац-ном состоянии равна
ск-ти роста цепи:![]() .
.
После
подставления в это ур-ние конц-ции
растущих рад-лов получим:
![]() (1)
(1)
В
стац-ном режиме отношение
![]() - константа
ск-ти полим-ции,
тогда
- константа
ск-ти полим-ции,
тогда
![]() .
.
След-но, ск-ть рад-ной полим-ции в массе пропорциональна конц-ции м-ра в первой степени и конц-ции инициатора в степени 0,5.
Константы kи находят по данным процесса в стац-ном состоянии. Анализ ур-ния (1) позволяет оценить влияние некоторых параметров процесса рад-ной полим-ции на ее ск-ть и размеры образ-ся цепных мол-л. vр представляет собой число мол-л м-ра, присоед-ся к растущим полимерным рад-лам в ед. времени. vоб опред-ся числом макрорад-лов, прекращающих рост в рез-те обрыва в ед. времени.
Отношение
vр/
vоб=
ν наз. Кинетической длиной цепи,
оно показывает, сколько мол-л м-ра
присоед-ся к растущему рад-лу до момента
прекращения его существования.
 ,
а после подстановки [R˙]:
,
а после подстановки [R˙]:
 .
.
Для
исключения из ур-ния частных констант
процесса полим-ции умножим числитель
и знаменатель на
![]() и с учетом того, что
и с учетом того, что
![]() ,
получим:
,
получим:
 .
.
Длина
кинетической цепи и ск-ть полим-ции
связаны между собой обратно пропорц-ной
зависимостью:
![]() .
Длина кинетической цепи в отсутствие
р-ций передачи цепи связана со средней
степенью полим-ции образ-ся макромол-л
.
Длина кинетической цепи в отсутствие
р-ций передачи цепи связана со средней
степенью полим-ции образ-ся макромол-л
![]() :
в случае обрыва цепи
:
в случае обрыва цепи
![]() ,
а при рекомбинации
,
а при рекомбинации
![]() или:
или:
--
при диспропорционировании -
![]()
![]() ,
,
--
при рекомбинации -
![]()
![]() .
.
На
начальной стадии полим-ции в массе
конверсия невелика и [M]=const.
Тогда из последних ур-ний следует, что
ММ пол-ра обратно пропорц-на
![]() ,
т.е. изменением [In]
можно регулировать ММ. Анализ кинетического
ур-ния рад-ной полим-ции позволяет также
оценить влияние тем-ры на общую ск-ть
процесса и размер образ-ся цепей. С
повышением тем-ры ск-ти всех трех
элементарных стадий полим-ции возрастают,
но не в равной мере. В силу различий
энергии активации каждой стадии тем-рные
коэффициенты р-ций иниц-ния, роста и
обрыва цепи различны: с повышением
тем-ры vи
возрастает
в
большей степени,
чем
vи
и vоб.
Т.к. vи=kи[In],
получим:
,
т.е. изменением [In]
можно регулировать ММ. Анализ кинетического
ур-ния рад-ной полим-ции позволяет также
оценить влияние тем-ры на общую ск-ть
процесса и размер образ-ся цепей. С
повышением тем-ры ск-ти всех трех
элементарных стадий полим-ции возрастают,
но не в равной мере. В силу различий
энергии активации каждой стадии тем-рные
коэффициенты р-ций иниц-ния, роста и
обрыва цепи различны: с повышением
тем-ры vи
возрастает
в
большей степени,
чем
vи
и vоб.
Т.к. vи=kи[In],
получим:
 .
.
След-но
увеличение vи
приводит к повышению общей ск-ти
полим-ции. Вместе с тем рост vи
приводит к повышению vи
и vоб
. Ск-ть обрыва цепи с повышением тем-ры
возрастает в большей степени( -[R˙]2).
Но т.к. средняя степень полим-ции образ-ся
макромол-л равна
![]() (a=1
или 2), то очевидно, что с ростом тем-ры
ср. коэффициент полим-ции уменьшается.
В случае фотохим-кой и радиационной
полим-ции ск-ть процесса в меньшей
степени зависит от тем-ры, чем при
инициир-нии процесса вещественными
инициаторами или нагреванием, а опред-ся
интенсивностью облучения.
(a=1
или 2), то очевидно, что с ростом тем-ры
ср. коэффициент полим-ции уменьшается.
В случае фотохим-кой и радиационной
полим-ции ск-ть процесса в меньшей
степени зависит от тем-ры, чем при
инициир-нии процесса вещественными
инициаторами или нагреванием, а опред-ся
интенсивностью облучения.
2.Стадия прекращения роста цепи вкл. два основных типа процессов: хим-кую дезактивацию концевых ФГ (потеря способ-ти к росту цепи); превращения, приводящие к образ-нию неактивных групп в данных условиях (при изменении условий полик-ции способ-ть к росту цепи возобновляется).
Дезактивация реакц-ных центров чаще всего закл-ся во взаимодействии их с монофункц-ными соединениями:

Концевая
бензоатная гр. не способна к дальнейшему
наращиванию цепи. Если долю вступивших
в р-цию с монофункц-ным соединением ФГ
обозначить через р', то при полном
расходовании этих групп (глубина
превращения р=1) их доля, израсходованная
на образование цепных мол-л, будет равна
(1- р'). Подставив это вырадение вместо р
в ур-ние Карозерса, получим
![]() ,
или
,
или
![]() ,
т.е. степень полик-ции пол-ра обратно
пропорц-на кол-ву монофункц-ного
соединения, вступившего в р-цию. Это
ур-ние верно при р=1. Если же р<1, то
,
т.е. степень полик-ции пол-ра обратно
пропорц-на кол-ву монофункц-ного
соединения, вступившего в р-цию. Это
ур-ние верно при р=1. Если же р<1, то
![]() .
.
Если
при полик-ции протекает несколько
параллельных р-ций дезактивации и их
доля в общей завершенности процесса
составляет
![]() , то суммарная относ-ная доля этих р-ций
равна
, то суммарная относ-ная доля этих р-ций
равна
![]() при полном расходовании ФГ.
при полном расходовании ФГ.
![]() или
или![]() - эти ур-ния универсальны и применимы
для большого числа р-ций прекращения
роста цепи.
- эти ур-ния универсальны и применимы
для большого числа р-ций прекращения
роста цепи.
Дезактивация ФГ может происходить не только при р-ции с монофункц-ными соединениями, но и вследствие циклизации с участием концевой группы или какого-либо ее побочного превращения. При полик-ции гексаметилендиамина и янтарной к-ты в расплаве рост цепи прекращается из-за внутримолекулярной циклизации:

Для процессов полик-ции с участием дикарбоновых к-т при повышенных тем-рах характерно декарбоксилирование: ~E-COOH → ~RH + CO2↑
При взаимодействии дифенолятов двухатомных фенолов с дихлорангидридами дикарбоновых к-т могул протекать побочные р-ции гидролиза хлоридных гр. с образ-нием карбоксильных гр., неактивных в данных условиях:

Прекращение роста цепи при активных ФГ обусловлено следующими основными причинами:
1) Достижение термодинамического равновесия (для обратимых реакций), когда конц-ция ФГ соответствует константе полик-ции. Изменяя Т, Р, можно изменить Кр, а след-но и конц-цию концевых гр., и ММ пол-ра;
2)Наличие в системе однотипных неактивных гр., возникющих из-за неэквимолярности исходных м-ров. Если м-р а-а взят в избытке по отношению к м-ру b-b, то пол-р содержит только концевые гр. а. Введя доп-но м-р b-b, можно увеличить ММ пол-ра;
3) Понижение активности ФГ за счет расходования кат-ра или понижения тем-ры Т полик-ции возобновляется;
4) воздействие физ. факторов (повышение вязкости, выпадение пол-ра в плотный осадок, образ-ние простр-ной сетки), делающие ФГ неподвижными или недоступными.
3. Исп-ние р-ля резко снижает вязкость рекц-ной системы и исключает диффузионные ограничения во взаимод-вии ФГ на завершающих стадиях процесса. Это приводит к повышению как ск-ти процесса, так и глубины превращения и способствует образ-нию более высокомол-ных продуктов. По сравнению с полик-цией в расплаве улучшается теплообмен, искл-ся местные перегревы реакционной смеси и связанные с ними деструктивные процессы. Но при полик-ции в р-ре есть и недостатки:
а) побочные р-ции с участием р-ля
б) меньшая эф-ность использования объема реакционной аппаратуры
в) наличие доп-ных стадий осаждения пол-ра, регенерации и очистки р-ля.
Этот метод занимает в промыш-ти 2-ое место. Ряд пол-ров может быть синтезирован и переработан только в р-ре (ароматические полиамиды, аром. полиэфиры, полиимиды). Н-р, поли-n-фенилентерефталамид получают полик-цией n-фениледиамина и дихлорангидрида ТФК в среде N,N'-диметилацетамида с добавлением неорг. солей:

Назначение р-ля при полик-ции в р-ре сводится к растворению исх. м-ров и образ-ся пол-ра, к облегчению удаления побочного соединения (отгонка в виде азеатропа, хим. связывание), к ускорению основной р-ции ступенчатого роста цепей, к изменению в соотношении р-ций роста цепи и циклизации.
При полик-ции с выделением воды в качестве НМС часто используют р-ли, образующие с ней низкокип. азеотропные смеси. Вода удаляется быстро и более полно, а равновесие смещ-ся в сторону образования более высокомол-ного пол-ра – азеотропная полик-ция.
В р-циях дихлорангидридов дикарбоновых к-т с диаминами веделяющийся HCl связывают амидными р-лями (диметилформамид, диметилацетамид), а также третичными аминами (пиридин,триэтиламин). В их отсутствии HCl вступает в реакцию с исх-ми диаминами с образ-нием нереакционноспособных или малоактивных дигидрохлоридов.
Природа р-ля сложным образом связана со ск-тью основной р-ции роста цепи при полик-ции: обычно ск-ть этого процесса опред-ся полярностью р-ля, его сольватирующей способностью и термодинамическим качеством по отношению к образ-ся пол-ру. Влияние среды чаще изучают на соотв-щих модельных р-циях НМС. Но при использовании данных о ск-тях модельных р-ций нужно учитывать возможность уменьшения константы ск-ти соотв-щей р-ции образ-ния макромол-л. Это происходит как вследствие повышения вязкости р-ра, так и в тех случаях, когда используемый р-ль является термодинамически плохим – макромол-лы в нем сворачиваются в плотные клубки, внутри которых могут оказаться концевые ФГ, не способные к последующим р-циям роста цепей из-за диффузионных ограничений. На рис представлена зависимость константы скорости синтеза поликарбоната от диэлектрической проницаемости смешенного растворителя:

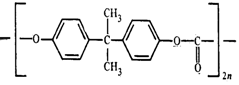

Прямые 1 и 2 отличаются лишь природой 2-го ком-та в смеси р-лей: дихлорбензол понижает только ɛ среды и не влияет на конформационное состояние мол-лы в р-ре: декалин понижает ɛ, но одновременно, являясь осадителем для п-ра, способствует сворачивание его макромол-л в клубки, плотность которых возрастает с увеличением доли декалина в смеси. Резко уменьшается ск-ть полик-ции – при таком же значении ɛ в смеси с n-дихлорбензолом она на 1,5 порядка выше. В тоже время ск-ть рции полимеробразования остается ниже ск-ти модельных р-ций НМ в-в, причем на последнюю не влияет природа р-лей. С понижением конц-ции м-ра в р-ре возрастает доля р-ций циклизации из-за уменьшения вероятности межмолекулярных взаимод-вие ФГ. Но в завис-ти от природы р-ля доля р-ций циклизации может существенно изменяться. Р-ль может изменять константу полик-ного равновесия, а также интенсивность побочных р-ций м-ров.
Влияние тем-ры. В зав-ти от тем-рного режима полик-цию в р-ре условно подразделяют на низкотем-рную (до 100°С) и высокотем-рную (выше 100°С). При сравнительно низких тем-рах осуществляют взаимод-вие дихлорангидридов дикарбоновых к-т с диаминами, дифенолами в присутствии акцепторов HCl , амидами, аминами и др.
Влияние тем-ры в более узких интервалах может быть разнообразным: ММ пол-ра может уменьшаться или увеличиваться с повышением тем-ры; иногда наблюдаются случаи экстремальной завис-ти (рис1)
Бимодальные завис-ти типа приведенной на рис1 обусловлены протеканием трех различных по мех-му р-ций роста цепей: некат-кой, кат-кой за счет основного катализа, кат-кой за счет нуклеофильного катализа. Изменение соотношения этих трех реакционных потоков и обуславливает сложную завис-ть приведенной вязкости полимера от тем-ры. Эта завис-ть аналогична тем-рной завис-ти между ск-тью полик-ции и ММ образ-ся полимера.
Высокотем-рную полик-цию используют в случае м-ров с относительно низкой реакционной способностью.


Билет 12
1. Полимеризация в растворе позволяет устранить главный недостаток блочной полимеризации – местные перегревы, поскольку выделяющаяся теплота идет на нагревание и частичное испарение растворителя, а также легче отводится вследствие лучшего перемешивания менее вязкой реакционной массы. Т.к. средняя степень полимеризации образующегося полимера пропорциональна концентрации мономера: а при разбавлении растворителем величина [M] уменьшается, то при полимеризации в растворе следует ожидать образования полимера меньшей ММ, чем при полимеризации в массе. Одновременно падает и скорость полимеризации: . При полимеризации в растворе может уменьшаться вследствие передачи цепи через растворитель. Полимеризацию в растворе по радикальному механизму в промышленности используют для синтеза ПВА, ПАН в N,N'-диметилформамиде или в водных растворах роданидов. Основной причиной, ограничивающей применение полимеризации в р-ре, кроме отмеченных, является необходимость использования растворителей( часто высокой чистоты) и их регенерации, а также затруднения при выделении полимера из р-ра.
2. Совместная поликонденсация (сополиконденсация) — разновид-ность полик-ции, при которой в образовании макромолекул участвует несколько мономеров (сомономеров). Образующийся при сополиконденсации полимер состоит из различных мономерных ос-татков и является поэтому сополимером. Естественно, что кроме молекулярной массы и характера ММР цепи такого сополимера должны характеризоваться и рас-пределением составных повторяющихся звеньев вдоль цепи.
Состав поликонденсационного сополимера, как и в случае цеп-ных процессов, определяется скоростью вступления сомономеров в полимерную цепь. Рассмотрим сополиконденсацию на примере двух оксикислот НОАСООН и НОВСООН с различной реакционной способностью окси- и карбоксильных групп. При образовании сопо-лимера протекают четыре реакции:
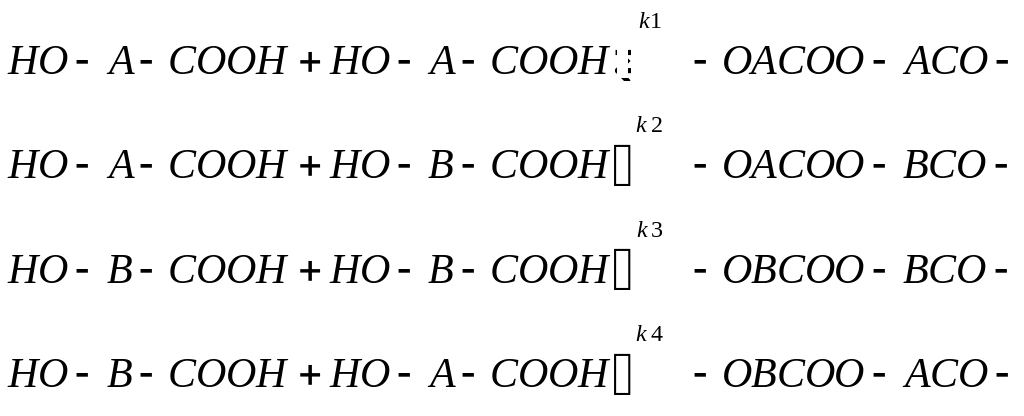
-Мономерные остатки А войдут в цепь за счет первой, второй и четвертой реакций, а остатки В — за счет второй, третьей и четвер-той реакций. Число мономерных звеньев А', вступивших в полимер-ную цепь, равно уменьшению количества мономера А в реакционной смеси:
 (1)
(1)
где [А] и [В] — концентрации соответствующих мономеров в реак-ционной смеси. Аналогичное выражение можно записать и для мо-номерных остатков В'. При делении этих выражений получают урав-нение, связывающее состав сополимера с составом мономеров в ис-ходной смеси:
 (2)
(2)
Уравнение (2) является дифференциальным, так как опре-деляет соотношение мономеров, вступивших в цепь в данный, сравнительно малый промежуток времени.
В рассматриваемом примере, как и в общем случае, при одина-ковом содержании мономерных звеньев А и В возможны различные варианты их соединения в цени:
—А — В — А — В — А — В — А — В — регулярно чередующийся сополимер
— Л — А — А — А — В — В — В — В — блок сополимер
— А — В — В — А — Л — Л — В — Л — статистический сополимер
Одним из способов оценки строения цепи является характер сое-динения двух звеньев. В этом способе наименьшим элементом, ко-торый характеризует строение макромолекулы, является диада - -двухзвенный фрагмент полимерной цепи. В рассматриваемом слу-чае такими диадами являются ~A—А~, ~ В—В~ и ~А—В~, т. е. две гомодиады (—А—А— и —В—B— ) и одна гетеродиада ( —А—В— ).
Строение цепей сополимера характеризуется относительной до-лей диад каждого вида; это осуществляют с помощью коэффици-ента микрогетерогенности Вм:

где,
![]() — доли
диад указанного состава.
— доли
диад указанного состава.
Для
гомополимера
![]() и,
следовательно, Вм=О. Для регуляр-но
чередующегося сополимера
и,
следовательно, Вм=О. Для регуляр-но
чередующегося сополимера
![]() и Вм=2. Статическому сополимеру отвечает
значение Вм=1. Когда преобладает
чередова-ние
звеньев Bм>1,
а
при преимущественно блочном строении
Вм<1.
и Вм=2. Статическому сополимеру отвечает
значение Вм=1. Когда преобладает
чередова-ние
звеньев Bм>1,
а
при преимущественно блочном строении
Вм<1.
Наиболее типичными случаями сополиконденсации является взаимодействие двух мономеров, из которых каждый в отдельности не способен образовать полимера (гетерополиконденсация мономе-ров а—а и b—b), а также интер-би-сополиконденсация (реакция двух мономеров а—а и одного b—b при эквивалентном соотношении реагирующих функциональных групп).
Поликонденсация
мономеров а—а и b—b,
примеры ко-торой
приведены выше, является простейшим
случаем сополикон-денсации,
для которого
![]()
![]() и
и
![]() .
По-этому
в уравнении (2) имеем А'/В'=1 или А'= В'. Это
означает, что образующийся сополимер
состоит из эквимолярных количеств
звеньев А и В со строгим их чередованием
—А—В—, так как обра-зование
гомодиад —А—А—
и —В—B
— здесь невозможно(
.
По-этому
в уравнении (2) имеем А'/В'=1 или А'= В'. Это
означает, что образующийся сополимер
состоит из эквимолярных количеств
звеньев А и В со строгим их чередованием
—А—В—, так как обра-зование
гомодиад —А—А—
и —В—B
— здесь невозможно(![]() и
)
.
и
)
.
Интер-би-сополиконденсации — является еще одним простым и важным методом получения сополимеров в ступенча-тых процессах; это поликонденсация двух однотипных мономеров для гетерополиконденсации (сомономеры типа а—а) с третьим (интермономером типа b —b), например:

Этот тип сополиконденсации характеризуется двумя константа-ми скорости образования макромолекул

а
состав сополимера выражается соотношением
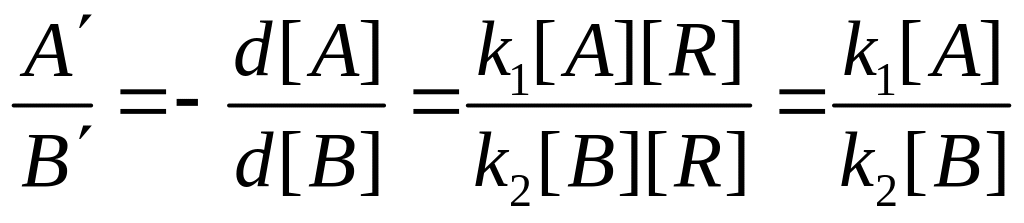 .
Обозначив
.
Обозначив
![]() и заменив текущие концентрации [А] и [В]
на [А]= [А]o
— [А' ] и [В] = [B]o — [В' ], после интегрирования
полу-чим
и заменив текущие концентрации [А] и [В]
на [А]= [А]o
— [А' ] и [В] = [B]o — [В' ], после интегрирования
полу-чим
 ,
где [A]o
и [B]o
— исходная концентрация мономеров А и
В; [А' ], [В' ] — концентрации мономерных
звеньев А и В, вступивших в по лимерную
цепь. На рис. представлены результаты
сополикон-денсации
дихлорангидрида изофталевой кислоты
со смесью n-фе-нилендиамина
и диолов, в качестве которых использованы
малоак-тивный
гидрохинон и его активное
бис-(β-гидроксиэтильное) производное:
,
где [A]o
и [B]o
— исходная концентрация мономеров А и
В; [А' ], [В' ] — концентрации мономерных
звеньев А и В, вступивших в по лимерную
цепь. На рис. представлены результаты
сополикон-денсации
дихлорангидрида изофталевой кислоты
со смесью n-фе-нилендиамина
и диолов, в качестве которых использованы
малоак-тивный
гидрохинон и его активное
бис-(β-гидроксиэтильное) производное:

А — n-фенилиденамин;
В — 1 — n-фенилендиамин;
— n-фенилиденамин;
В — 1 — n-фенилендиамин;
2— бис-(β-гидрокси)гидрохинон (активный сомономер); 3 — гидрохинон (малоактивный сомоно-мер)
где Х—просто связь (гидрохинон) или группа СН2СН2О (бис-β- гидроксиэтилгидрохинон) .
При замене части n-фенилендиамина на относительно активный , сомономер (бис-β-гидроксиэтилгидрохинон) кривая состава сопо-лимера мало отличается от кривой состава сополимера, полученно-го без использования сомономера В ( кривая 2). В слу-чае малоактивного сомономера В (гидрохинон) сополимер оказы-вается обедненным звеньями этого сомономера, при этом сущест-венно понижается и ММ сополимера.
Строение цепей сополимеров, образующихся при интер-би-сопо-ликонденсации, характеризуют по относительному содержанию триад ~ARAR~, ~ARBR~ и ~BRBR~, которые по сути иден-тичны диадам.
3. Для получения пол-ров с max ММ необходимо проводить полик-цию при эквимольном соотношении ФГ м-ров. Наиболее просто это достигается по конденсации м-ров типа a-b (a=b), например аминокарбоновая к-та. Но если есть примеси монофункц. групп, баланс ФГ нарушается и длина цепей уменьшается. Поэтому важно знать кол-ную связь между избытком ФГ одного вида и ММ пол-ра.
Рассмотрим
полик-цию м-ра а-а с избытком м-ра b-b.
Число ФГ равно Na
и Nb
соотв-но. Оно вдвое больше числа мол-л
в системе. Стехиометрический разбаланс
выражается как
![]() Суммарное число мол-л в системе
Суммарное число мол-л в системе
![]() или
или
![]()
Если
в даны пром-к времени доля прореагировавших
групп а составила р, то доля прореагир-ших
групп b
составила rp,
а не прореагировавших групп aи
b
будет равна (1-р) и (1-rp).
Общее число концевых групп равно сумме
непрореагар-ших групп а и b,
а суммарное число макромол-л равно 0,5
общего кол-ва концевых групп:
![]() .
Поскольку среднечисловая степень
полик-ции
.
Поскольку среднечисловая степень
полик-ции
![]() равна общему числу мол-л а-а и b-b
в системе, деленному на число мол-л
пол-ра, то:
равна общему числу мол-л а-а и b-b
в системе, деленному на число мол-л
пол-ра, то:
 .
Это ур-ние показывает, как изменяется
в
зав-ти от стехиометрического разбаланса
r
и ст. завершенности р-ции p.
При r=1
оно переходит в ур-ние Карозерса
.
Это ур-ние показывает, как изменяется
в
зав-ти от стехиометрического разбаланса
r
и ст. завершенности р-ции p.
При r=1
оно переходит в ур-ние Карозерса
![]() ,
а в случае
,
а в случае
![]() имеем
имеем
![]() .
Справедливость последнего выр-ния
многократно подтверждена экспер-но для
просессов как с выделением НМС(рис.1),
так и без его выделения(рис2)
.
Справедливость последнего выр-ния
многократно подтверждена экспер-но для
просессов как с выделением НМС(рис.1),
так и без его выделения(рис2)

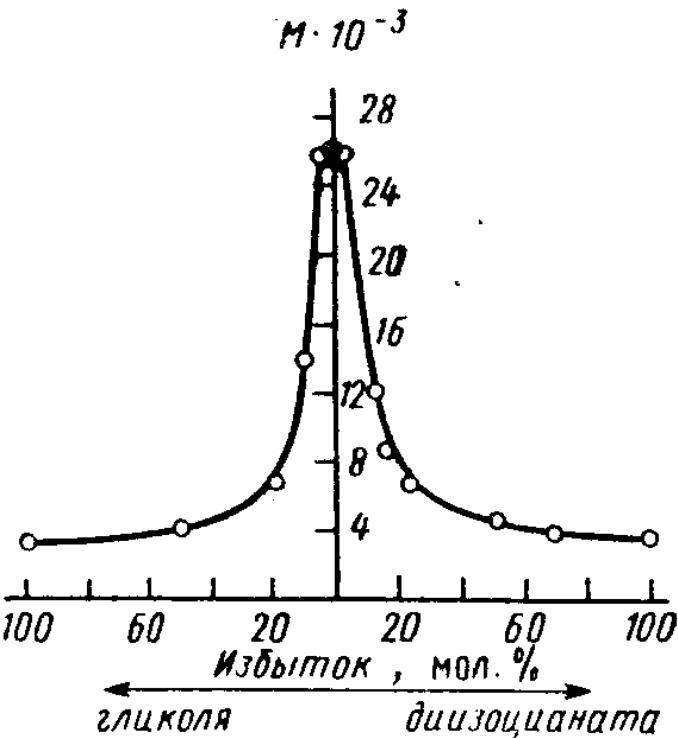
Рис. подтверждают, что max ММ достигается при равномолярном соотношении исх. м-ров а-а и b-b. Но иногда максимум ММ смещен в сторону избытка одного из м-ров. Это бывает, когда один из типов ФГ участвует в побочных р-циях, что изменяет баланс реакционных центров и делает необходимым его восстановление введением избытка этих групп.
Использование избытка одного из сом-ров – Эффективный метод синтеза олигомеров с определенной ММ и концевыми группами одного типа. При этом max ММ достигается также при эквимольном соотношении ФГ. Последнее ур-ние справедливо и для гетерополик-ции м-ра a-b или сом-ров a-a и b-b в присутствии монофункц-ного соединения, например для полик-ции диолов и дикарбоновых к-т присутствии монокарбоновой к-ты:

или для поликонд-ции дикарб. к-ты с диамином в присутствии моноамина:

Стехиометрический
разбаланс определяют в общем виде по
![]() ,
где
,
где
![]() -
число мол-л монофунц-ного соединения в
системе.
-
число мол-л монофунц-ного соединения в
системе.
Для
поликонц-ции м-ра a-b
в присутствии монофункц-ного соединения
b'
величину r
находят
![]() ,
где Nab=Na=Nb
и равно числу мол-л м-ра a-b
,
где Nab=Na=Nb
и равно числу мол-л м-ра a-b
Билет 13
Радикальная полимеризация в эмульсии – наиболее распространенный промышленный метод синтеза полимеров радикальной полимеризацией. Преимущества: большая скорость процесса; возможность его проведения при более низкой температуре, более высокая молекулярная масса и узкое ММР полимера.
Реакционная система содержит: дисперсионную среду (деминерализованная вода); мономер (30-60% от массы вода); эмульгатор (ПАВ – соли щелочных металлов и жирных кислот (мыла), соли сульфокислот); инициатор радикальной полимеризации, растворимый в воде (пероксид водорода, персульфат Na+, K+, NH4+), регуляторы и буферные вещества для поддержания постоянным значение pH среды.
Э мульгаторы
частично растворимы в воде, но выше
некоторой критической концентрации
мицеллообразования образуют агрегаты
молекул эмульгатора сферической или
чаще стержнеобразной формы. Внутренняя
часть мицеллы образована гидрофобными
УВ фрагментами молекул эмульгатора, а
наружная – гидрофильными. Обычно
содержание эмульгатора – 1-5% от массы
мономера. Мономер, нерастворимый или
растворимый в воде, находится в виде
капель размерами 1-10 мкм, размер мицелл
эмульгатора – в 100 раз меньше. С увеличением
содержания эмульгатора в системе
увеличивается число мицелл меньших
размеров, т.е. увеличивается их удельная
поверхность. Большая часть мономера
находится в виде капель: их число 1010
– 1011 в 1 мл, а мицелл около 1018
частиц в 1 мл. Т.к. концентрация мономера
в воде мала, то полимеризация в водной
фазе не идет. Невозможна полимеризация
и в каплях мономера, т.к. в нем не растворим
инициатор. Полимеризация идет только
в мицеллах, содержащих мономер. Радикалы
инициатора из водной фазы проникают в
мицеллы, где и начинается рост цепи. По
мере протекания полимеризации высокая
концентрация мономера в мицеллах
поддерживается за счет его диффузии из
капель, при этом мицеллы растут.
Полимеризация инициируется только
~0,1% мицелл, присутствующих в смеси.
Активные мицеллы растут, захватывая на
свою поверхность все больше молекул
эмульгатора из смеси, понижая его
концентрацию ниже предела мицеллообразования,
способствуют растворению неактивных
мицелл. При конверсии 2-15% размер активных
мицелл становится существенно больше
размера исходных мицелл; поэтому активные
мицеллы называют полимерно-мономерными
частицами (ПМЧ). Число ПМЧ в процессе
полимеризации не меняется. При конверсии
50-80% капельки мономера полностью исчезают,
а еще не прореагировавший мономер
содержится в ПМЧ. Размер ПМЧ на завершающей
стадии процесса составляет 0,05-02 мкм
(промежуточное положение между исходными
мицеллами и капельками мономера
(см.рис.).
мульгаторы
частично растворимы в воде, но выше
некоторой критической концентрации
мицеллообразования образуют агрегаты
молекул эмульгатора сферической или
чаще стержнеобразной формы. Внутренняя
часть мицеллы образована гидрофобными
УВ фрагментами молекул эмульгатора, а
наружная – гидрофильными. Обычно
содержание эмульгатора – 1-5% от массы
мономера. Мономер, нерастворимый или
растворимый в воде, находится в виде
капель размерами 1-10 мкм, размер мицелл
эмульгатора – в 100 раз меньше. С увеличением
содержания эмульгатора в системе
увеличивается число мицелл меньших
размеров, т.е. увеличивается их удельная
поверхность. Большая часть мономера
находится в виде капель: их число 1010
– 1011 в 1 мл, а мицелл около 1018
частиц в 1 мл. Т.к. концентрация мономера
в воде мала, то полимеризация в водной
фазе не идет. Невозможна полимеризация
и в каплях мономера, т.к. в нем не растворим
инициатор. Полимеризация идет только
в мицеллах, содержащих мономер. Радикалы
инициатора из водной фазы проникают в
мицеллы, где и начинается рост цепи. По
мере протекания полимеризации высокая
концентрация мономера в мицеллах
поддерживается за счет его диффузии из
капель, при этом мицеллы растут.
Полимеризация инициируется только
~0,1% мицелл, присутствующих в смеси.
Активные мицеллы растут, захватывая на
свою поверхность все больше молекул
эмульгатора из смеси, понижая его
концентрацию ниже предела мицеллообразования,
способствуют растворению неактивных
мицелл. При конверсии 2-15% размер активных
мицелл становится существенно больше
размера исходных мицелл; поэтому активные
мицеллы называют полимерно-мономерными
частицами (ПМЧ). Число ПМЧ в процессе
полимеризации не меняется. При конверсии
50-80% капельки мономера полностью исчезают,
а еще не прореагировавший мономер
содержится в ПМЧ. Размер ПМЧ на завершающей
стадии процесса составляет 0,05-02 мкм
(промежуточное положение между исходными
мицеллами и капельками мономера
(см.рис.).
После полной адсорбции эмульгатора на поверхности ПМЧ образование новых частиц прекращается и скорость полимеризации становится постоянной, т.к. скорость диффузии мономера из капель ПМЧ значительно превышает скорость его полимеризации в ПМЧ. Число ПМЧ в 1 мл составляет 1013 – 1015 частиц и является постоянным. В каждой ПМЧ не более одного радикала – иначе происходит обрыв цепи.
При попадании инициирующего радикала в ПМЧ идет рост цепи (если в ней не было растущего радикала) или ее обрыв (если в ПМЧ уже был растущий радикал). В любой момент половина ПМЧ активна (в них идет полимеризация), а вторая половина – неактивна. Скорость полимеризации равна:
![]()
где N – число ПМЧ.
С корость
полимеризации не зависит от скорости
инициирования, а определяется N.
Она будет постоянной, если концентрация
эмульгатора постоянна, однако она
изменяется при варьировании количества
ПАВ (см. рис.):
корость
полимеризации не зависит от скорости
инициирования, а определяется N.
Она будет постоянной, если концентрация
эмульгатора постоянна, однако она
изменяется при варьировании количества
ПАВ (см. рис.):
С увеличением концентрации эмульгатора скорость повышается, после достижения постоянства числа ПМЧ скорость также достигает постоянного значения.
Увеличение скорости полимеризации на начальной стадии связано с ростом числа ПМЧ, а понижение ее на конечной стадии - с уменьшением концентрации мономера.
Скорость ρ, с которой первичный радикал проникает в частицу, равна:
![]()
тогда
![]()
Скорость полимеризации совпадает с длиной кинетической цепи. В эмульсионной полимеризации скорость процесса и молекулярную массу полимера можно увеличить при постоянной скорости инициирования (rи = const) повышением числа ПМЧ, которое можно рассчитать:
![]()
где μ – скорость увеличения объема ПМЧ; ап – пограничная поверхность, занятая молекулами эмульгатора в мицеллах; [Э] – концентрация эмульгатора.
Часто эмульсионную полимеризацию осуществляют в присутствии ОВ систем, состоящих из персульфата и соли двухвалентного железа:

или гидроперекиси и соли двухвалентного железа:
ROOH + Fe+2 Fe+3 + HO- + RO*
Для предотвращения загрязнения полимера солями железа их используют в небольших количествах, вводя в систему восстановитель (глюкозу или фруктозу), переводящий обратно Fe+3 в Fe+2. При этом схему инициирования можно представить следующим образом:

ОВ системы позволяют снизить температуру полимеризации для нетермостойких мономеров.
Иногда эмульсионная полимеризация возможна без эмульгатора: инициирующие радикалы реагируют с мономером, частично растворенным в воде, образуя олигомер, выполняющий роль ПАВ (например гетерофазная безэмульгаторная полимеризация стирола, ВА).