

Глава 6. Системы переменного состава. Химический потенциал
6 - 1. Термодинамические функции систем переменного состава
В предыдущей главе характеристические термодинамические функции рассматривались для систем, состав которых оставался постоянным. Изменение состава может быть вызвано химической реакцией.
Если система содержит вещества А1,А2, ... , Аi, ..., числа молей которых соответственно равны n1, n2, ..., ni, ..., то в ходе реакции они должны измениться.
Системы, в которых переменными становятся числа молей веществ, называются системами переменного состава. Применяется также название системы с переменным числом частиц.
В этих системах, наряду с внешними параметрами и энтропией, от новых переменных зависят термодинамические функции, а именно:
внутренняя энергия становится функцией от объема, энтропии и чисел молей веществ
U = U(V,S,n1,n2, ...ni, ...),
энтальпия - функцией давления, энтропии и чисел молей
H = H(P,S,n1,n2, ..., ni, ...),
энергия Гельмгольца - функцией объема, температуры и чисел молей
F = F(V,T,n1,n2, ...,ni, ...),
энергия Гиббса - функцией давления, температуры и чисел молей
G = G(P,T, n1,n2, ....,ni, ... ).
Приращения этих функций с учетом влияния чисел молей оказываются равными
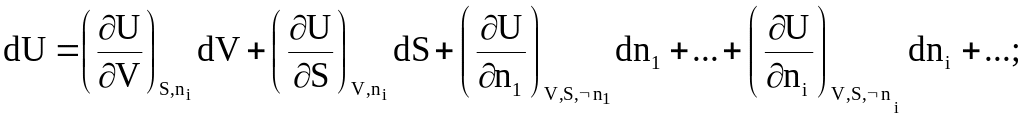
![]()
![]()
![]()
Символы n1, ..., ni, ... в данном случае означают, что постоянными остаются числа молей всех веществ, кроме стоящих за знаком .
С учетом частных производных термодинамических функций, которые подробно рассматривались в предыдущей главе, вышеприведенные уравнения в компактной форме записи принимают следующий вид:
![]() (6 - 1)
(6 - 1)
![]() (6 - 2)
(6 - 2)
![]() (6 - 3)
(6 - 3)
![]() (6 - 4)
(6 - 4)
6 - 2. Парциальные молярные величины
Входящие в уравнения (6 - 1) - (6 - 4) частные производные термодинамических функций относятся к числу парциальных молярных величин.
Парциальной молярной величиной экстенсивной характеристики вещества называют частную производную этой характеристики по числу молей данного вещества при постоянных числах молей других веществ и постоянных параметрах, включая энтропию, влияющих на эту характеристику.
Обычно парциальная молярная величина обозначается с помощью черты над соответствующей экстенсивной характеристикой с индексом, обозначающим вещество.
В соответствии с приведенным определением и системой обозначения в общем виде парциальная молярная величина экстенсивной характеристики Z вещества i выражается следующим образом:
![]() ,
(6 - 5)
,
(6 - 5)
где и - параметры системы, поддерживаемые постоянными.
В качестве примеров приведем один из способов нахождения парциального молярного объема.
Если необходимо найти парциальный молярный объем воды в растворе этого состава, то раствор следует поместить в емкость, позволяющую с высокой точностью измерять небольшие изменения объема. Чаще всего для этого используется колба , снабженная сверху капиллярной трубкой. Налив испытуемый раствор в сосуд, который после взвешивания помещается в термостат, можно определить исходный объем жидкости. Затем, добавив незначительный объем воды к этому раствору, после нового взвешивания и термостатирования можно найти новый объем жидкости. Пусть изменение объема раствора оказалось равным V, причем эта величина во много раз меньше объема раствора (V<<V), а масса добавленной воды равна mH2O. Тогда парциальный молярный объем воды можно рассчитать по формуле
 ,
,
в которой MH2O - молярная масса воды.
В рассмотренном примере для оценки парциального молярного объема воды достаточно было строго поддерживать постоянной только температуру, так как вода практически несжимаема и изменения атмосферного давления не могут повлиять на полученные результаты. Газы очень чувствительны к изменению давления. Поэтому для определения парциального объема газов системы следует тщательно маностатировать.
Чтобы найти парциальный объем газа в газовой смеси, последнюю взвешивают, термостатируют и маностатируют. Затем к смеси подкачивают небольшую порцию чистого газа, парциальный объем которого хотят определить, и смесь вновь взвешивают и маностатируют. После этого определяют изменение объема. Отношение изменения объема к числу молей добавленного чистого газа при постоянном давлении и постоянной температуре равно парциальному объему этого газа.
Отметим, что парциальные молярные величины интенсивных характеристик системы не имеют смысла. Вряд ли, например, кто-нибудь воспользуется понятием парциальной молярной температуры.
В связи с этим широко применяемый термин «парциальное давление» можно рассматривать как случайный.
6 - 3. Химический потенциал
Химическим потенциалом i называется парциальная молярная энергия Гиббса при постоянном давлении, постоянной температуре и постоянных числах молей остальных веществ
![]() (6 - 6)
(6 - 6)
С учетом равенства (6 - 6) уравнение (6 - 4) принимает следующий вид:
dG = VdP - SdT + idni. (6 - 7)
Так как энергия Гельмгольца может быть выражена
F = G - PV,
то ее приращение выразится так:
dF = dG PdV VdP,
dF = PdV SdT + idni. (6 - 8)
Для других функций
H = G + TS;
U = G PV + TS
найдем
dH = VdP + TdS + idni, (6 - 9)
dU = PdV + TdS + idni. (6 - 10)
Следовательно, химический потенциал можно выразить как парциальную молярную величину любой характеристической термодинамической функции:
![]() (6 - 11)
(6 - 11)
![]() (6 - 12)
(6 - 12)
![]() (6 - 13)
(6 - 13)
![]() (6 - 14)
(6 - 14)
Введенный Дж. У. Гиббсом химический потенциал оказался очень удобной для термодинамических расчетов величиной.
Рассмотрим одно из важнейших свойств химического потенциала.
Пусть находящаяся в равновесии система состоит из фаз, которым присвоим номера 1,2. ... (фазой называются части системы, имеющие одинаковый состав и свойства в каждой точке). Во всех фазах содержится вещество i.
Данное условие поясним следующими примерами.
В равновесии с паром находится водный раствор этанола. В этом случае в равновесии находятся две фазы, одна из которых жидкая (раствор), а другая газообразная (пар). Этанол находится в обеих фазах.
Система состоит из двух слоев жидкости: верхний слой - бензол, а нижний слой - вода. В систему вводится кристаллический йод, который растворим как в воде, так и в бензоле. Через определенное время в системе устанавливается равновесие и обе жидкости содержат растворившийся в них йод.
В приведенных выше примерах этанол и йод являются теми веществами, которые находятся во всех фазах системы.
В равновесных системах все фазы имеют одинаковую температуру. Система находится также при постоянном давлении. Поэтому условием равновесия является минимум энергии Гиббса, т.е.
dG = 0.
Перенеся из фазы 1 в фазу 2 бесконечно малое число молей dni вещества i, изменим энергию Гиббса первой фазы на следующую величину:
dG(1) = i(1)(-dni).
Знак минус означает, что вещество i выводится из фазы 1. Верхний индекс у символа химического потенциала и энергии Гиббса показывает, что они относятся к первой фазе.
Изменение энергии Гиббса второй фазы составит
dG(2) = i(2)dni.
Так как энергия Гиббса обладает свойством аддитивности, то общее изменение энергии Гиббса в системе находим суммированием изменений в отдельных фазах
dG = dG(1) + dG(2) = i(1)(-dni) + i(2)dni = 0.
Следовательно,
i(1) = i(2).
Далее, перенося то же бесконечно малое число молей dni вещества i из фазы 2 в фазу 3 и т.д., получим остальную часть системы равенств:
i(2) = i(3), ..., i(-1) =i().
Окончательно условие фазового равновесия можно записать в следующей форме
i(1) =i(2) =...=i() . (6 - 15)
Уравнение (6 - 15) означает, что химический потенциал одного и того же вещества во всех равновесных фазах равен одной и той же величине.
Применение химического потенциала существенно упрощает рассмотрение химического равновесия.
Когда автор начинал преподавательскую деятельность, он слышал одну любопытную историю, в которой подчеркивалось значение работ Гиббса по введению химического потенциала. Много лет спустя в пересказе по памяти она выглядит следующим образом.
Джозайя Уиллард Гиббс, один из основоположников современной формы термодинамики, пришедшей на смену термодинамики циклов Томсона, Клаузиуса, Максвелла, родился в 1839 г. В первой половине ХХ в. термодинамика была наиболее популярной дисциплиной у физиков и химиков и столетие со дня рождения Гиббса в Москве отмечалось с большой торжественностью. Юбилейное заседание проводилось в самой крупной научной аудитории того времени - в Большой аудитории Политехнического музея. Основным докладчиком был переводчик на русский язык и комментатор работ Гиббса Владимир Ксенофонтович Семенченко. Слушателей поразили две вещи из его доклада. Говоря с восторгом о Гиббсе, В. К. Семенченко, чтобы еще больше подчеркнуть ту тщательность, с которой Гиббс подходил к отделке своих работ, с большим пафосом сказал: «Гиббс отличался необычайной научной добросовестностью, совершенно невиданной в наши дни». Акцентируя внимание на этих словах, он наклонился к президиуму, в котором сидели видные ученые и другие деятели. Получился весьма назидательный поклон. Продолжая свое выступление, он подошел к одному из крупнейших достижений Гиббса - введению в термодинамику химического потенциала. Эта часть доклада была выражена следующим образом. Последовала фраза: «Введя в термодинамику химический потенциал, Гиббс создал совершенную науку, можно сказать, науку для дураков!». Несомненно, читатель понял смысл этой фразы правильно, а именно: химический потенциал сделал термодинамику стройной и логичной наукой, в которой вопросы равновесия решаются приравниванием химических потенциалов.
Автор увидел В.К.Семенченко через несколько десятилетий после описываемых событий и был поражен эмоциональностью и образностью выступления уже достаточно немолодого человека. Поражало его стремление сделать свое выступление доступным каждому слушателю. Он все время апеллировал к слушателям, сопровождая свою речь жестами. Ему хотелось, чтобы каждый проникся основной идеей его выступления. Можно представить, какова же была его энергия и с каким темпераментом он делал доклад на том заседании!
Автор полагает, что эта анекдотическая история не исказит образ В.К.Семенченко, замечательного человека и ученого, создавшего теорию критических фазовых переходов, к которым относятся переходы металлов из ферромагнитного в парамагнитное состояние и др.
6 - 4. Уравнение Гиббса - Дюгема
Энергия Гиббса является функцией интенсивных величин Р и Т и чисел молей веществ, не влияющих на интенсивные величины. Таким образом, энергия Гиббса оказывается однородной функцией первой степени от чисел молей веществ.
Как известно, для однородных функций первой степени справедлива формула Эйлера, которая применительно к данному случаю может быть представлена следующим образом:
![]() (6 - 16)
(6 - 16)
или
![]() .
(6 - 17)
.
(6 - 17)
Полное приращение энергии Гиббса в соответствии с уравнением (6 - 17) дает
![]() .
(6 - 18)
.
(6 - 18)
Сопоставление уравнения (6 - 18) с уравнением (6 - 7), у которых одинаковы левые части, дает
![]() . (6 - 19)
. (6 - 19)
Уравнение (6 - 19) называется уравнением Гиббса - Дюгема.
Для постоянных Р и Т уравнение Гиббса - Дюгема приобретает следующую форму:
![]() (6 - 20)
(6 - 20)
6 - 5. Зависимость химического потенциала от состава идеальных газовых систем
Первыми идеальными системами, к которым мы обратимся, являются идеальные газовые смеси. Напомним, какими свойствами они обладают.
Смеси подчиняются уравнению состояния идеального газа
![]() ,
(6 - 21)
,
(6 - 21)
в котором ni - число молей газа i, и закону Дальтона, согласно которому общее давление смеси равно сумме парциальных давлений газов
![]() .
(6 - 22)
.
(6 - 22)
Для числа молей ni каждого газа выполняется уравнение
![]() .
(6 - 23)
.
(6 - 23)
При постоянной температуре для системы с постоянным объемом выполняются равенства:
![]() .
(6 - 24)
.
(6 - 24)
При постоянной температуре уравнение Гиббса - Дюгема (6 - 19) приобретает форму:
![]() .
(6 - 25)
.
(6 - 25)
Из равенства левых частей уравнений (6 - 24) и (6 - 25) следует
![]() .
.
или
![]() .
(6 - 26)
.
(6 - 26)
В идеальной смеси газы не влияют друг на друга (ведут себя независимо). В связи с этим разность в каждой скобке должна быть равна нулю:
![]() .
(6 - 27)
.
(6 - 27)
Напомним, что уравнение (6 - 27) выполняется при постоянной температуре. Его интегрирование дает
![]() .
(6 - 28)
.
(6 - 28)
В уравнении (6 - 28) для каждой температуры получается своя постоянная интегрирования. Для краткости записи ее чаще всего обозначают io. С учетом этого традиционная форма зависимости химического потенциала от парциального давления идеального газа приобретает следующий вид:
![]() (6 - 29)
(6 - 29)
При постоянном общем давлении и при постоянной температуре выполняется равенство
![]() .
.
Отношение числа молей данного вещества ni к общему числу молей ni является молярной долей вещества xi.
Введем обозначение
![]() ,
,
с помощью которого выразим зависимость химического потенциала газа от его молярной доли в идеальной газовой смеси
![]() . (6 - 30)
. (6 - 30)
6 - 6. Зависимость химического потенциала от состава идеальных растворов конденсированных систем
Конденсированными системами называются жидкие или твердые системы.
Идеальными растворами в таких системах считаются растворы, подчиняющиеся закону Рауля.
Ф.Рауль экспериментально исследовал давление пара над растворами. Результаты его исследований для многих растворов можно представить в следующей форме:
Давление пара вещества над идеальным раствором прямо пропорционально молярной доле этого вещества в растворе
![]() (6 - 31)
(6 - 31)
Обозначим давление пара над чистой жидкостью Pio (ее составу отвечает xil=1). С учетом этого уравнение, выражающее закон Рауля, принимает следующий вид:
![]() (6 - 32)
(6 - 32)
Примем, что пар над раствором подчиняется уравнению состояния идеального газа.
В равновесной системе химический
потенциал вещества i в
паре
![]() равен химическому потенциалу этого же
вещества в жидком растворе
равен химическому потенциалу этого же
вещества в жидком растворе
![]() .
Поэтому можно записать равенство
.
Поэтому можно записать равенство
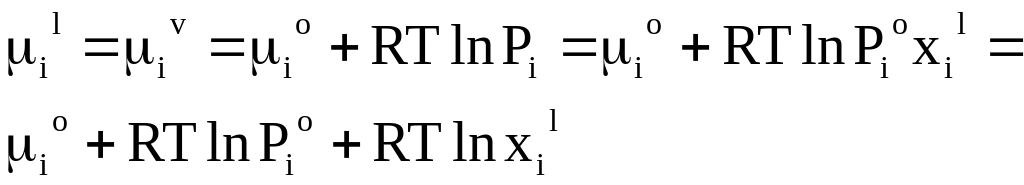
Давление насыщенного пара над чистым веществом при постоянной температуре является постоянной величиной.
Введем новую константу
![]() .
(6 - 33)
.
(6 - 33)
Зависимость химического потенциала вещества от его молярной доли в идеальном растворе выразится следующим образом:
![]() (6 - 34)
(6 - 34)
В уравнениях (6 - 29), (6 - 30) и (6 - 34) константы обозначались разными символами, чтобы подчеркнуть разнообразие типов зависимости химического потенциала от состава системы. В дальнейшем постоянную будем обозначать единым способом для всех случаев.
6 - 7. Реальные газовые системы. Летучесть
В предыдущих параграфах было показано, как, используя уравнение состояния идеального газа, была найдена зависимость химического потенциала вещества в идеальной смеси от содержания этого вещества (парциального давления или молярной доли).
Все идеальные газы подчиняются одному и тому же уравнению состояния. В отличие от них практически для каждого реального газа требуется свое уравнение состояния, отличающееся если не по форме, то входящими в него константами.
Например, в известном уравнении Ван-дер-Ваальса
![]()
константа а может изменяться в несколько раз, а отношение ее значений для октана и гелия превышает 1000.
Если использовать отдельные уравнения для нахождения зависимости химического потенциала от содержания вещества в смеси, то пришлось бы для каждого вещества создавать свою термодинамику, что сделало бы необычайно сложным практическое применение результатов.
Развитие термодинамики пошло по другому пути, в разработке которого огромную роль сыграли работы Льюиса и Рэндалла. Было предложено сохранить форму термодинамических уравнений, полученных для идеальных газовых смесей, заменив давление функцией от давления. Поскольку зависимость химического потенциала от содержания вещества в смеси применима только к условию постоянной температуры, то и вновь вводимая функция должна быть изотермической, т.е. устанавливаться каждый раз при изменении температуры.
Функция, заменившая давление в термодинамических уравнениях для реальных газов, получила название летучести, или фугитивности. Ее обычно обозначают f.
Условия введения летучести таковы:
при приближении состояния реального газа к свойствам идеального газа (это может происходить при понижении давления) летучесть приближается к давлению, что означает полное совпадение летучести и давления у идеальных газов;
форма уравнений с летучестью полностью повторяет форму уравнений, выведенных для идеального газа.
Сравним эти формы друг с другом и общей формой термодинамических уравнений в изотермических условиях (см. табл. 6 - 1).
Таблица 6 - 1
Сравнение форм термодинамических
уравнений для газов
|
Общая форма термодинамиче-ского уравнения |
Форма уравнения для идеального газа |
Форма уравнения для реального газа |
Бесконечно малое (элементарное) приращение энергии Гиббса |
|
|
|
Конечное приращение энергии Гиббса |
|
|
|
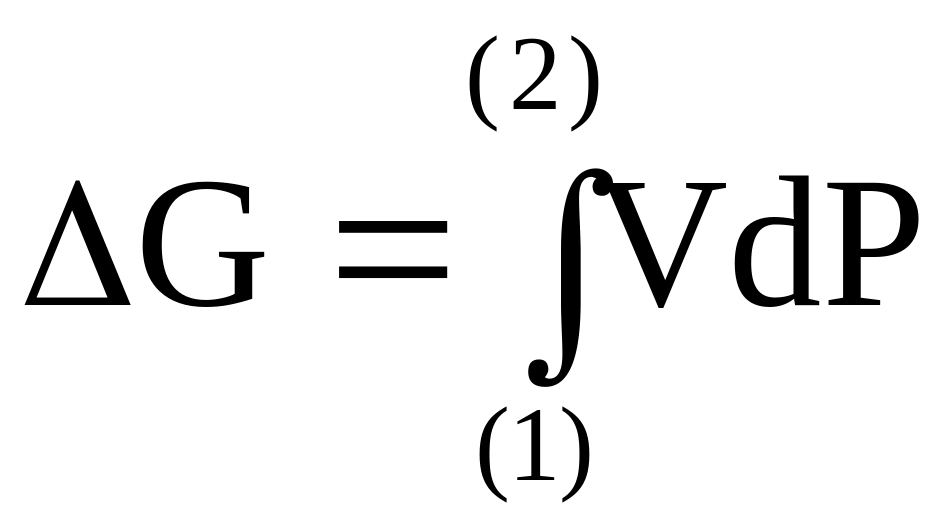
Приравнивая правые части уравнений в первом и последнем столбце последней строки, получим
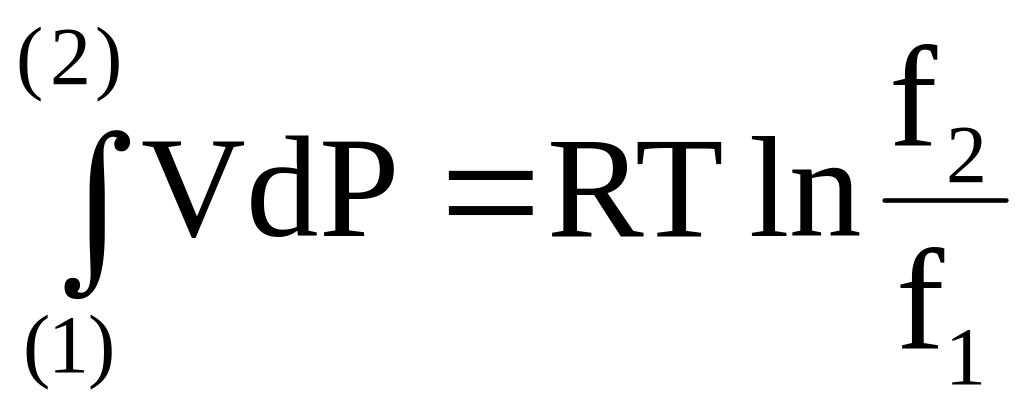 (6 - 35)
(6 - 35)
или
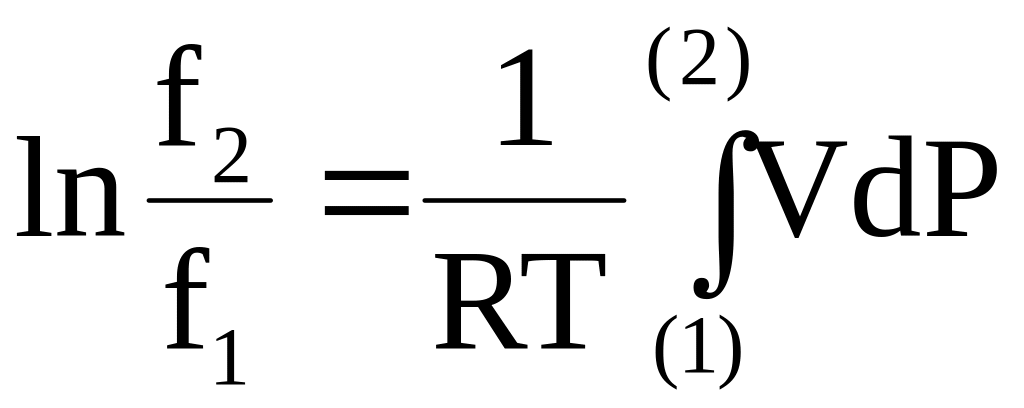 .
(6 - 36)
.
(6 - 36)
Уравнение (6 - 36) служит для нахождения отношения летучестей.
Обычно при заданной температуре находят зависимость объема газа от его давления и проводят интегрирование функции V=V(P) от давления Р1 до давления Р2. Интегрирование осуществляют или с применением эмпирического уравнения, отражающего зависимость объема от давления, или графическими методами на основе экспериментально полученного графика этой функции.
О проявляемом физико-химиками искусстве графического интегрирования можно судить по следующей истории.
Рассказывают, что выдающийся американский изобретатель Т.Эдисон в преклонном возрасте решил найти себе преемника. Был объявлен конкурс, в ходе которого Эдисон предложил молодым людям найти объем колбы электрической лампы. Молодые претенденты на звание преемника самого Эдисона предлагали разнообразные способы, включая проектирование колбы на большой экран, чтобы найти уравнение поверхности колбы для последующего интегрирования. К своему сожалению, Эдисон не нашел в числе предложенных ими ни одного простого и точного способа. После окончания конкурса он взвесил колбу, налил в нее воду и снова взвесил. Объем колбы был установлен с высокой точностью.
Мало кто из физико-химиков знает о конкурсе, устроенном Эдисоном. Однако методикой определения площади под кривой, т.е. графического интегрирования, владеют многие из них. Когда необходимо определить площадь под кривой, физико-химик проводит эту кривую на миллиметровой бумаге, а затем вырезает ножницами из нее фрагмент, содержащий отрезок по абсциссе, два отрезка, перпендикулярных оси абсцисс и касающихся начала и конца кривой, и саму кривую. Затем взвешивает лист миллиметровой бумаги, площадь которого известна. Читатель может сам оценить точность такого интегрирования, если учесть, что масса вырезанного фрагмента близка к 5 г, а точность взвешивания на обычных аналитических весах составляет 0,0002 г.
Измерения и расчеты, проводимые по уравнению (6 ‑ 36), дают возможность находить только отношение летучестей. Абсолютное значение летучести отдельного газа при заданном давлении установить, пользуясь только этим уравнением, невозможно. Таким образом, одного уравнения для создания таблиц значений летучести, которые позволили бы накапливать экспериментальные данные для последующего применения, оказалось недостаточно. Потребовалось ввести дополнительные условия для представления летучести в виде справочной величины. Таким дополнительным условием было установление точки начала отсчета летучести.
6 - 8. Стандартное термодинамическое состояние для газов
Стандартное термодинамическое состояние было введено как общее начало отсчета летучести для всех газов.
Так как свойства всех газов различны, то в реальных условиях у них не может быть общих точек на кривой f=f(P). Следовательно, общее для всех газов состояние может быть только воображаемым.
Наиболее удобно предполагать, что все свойства у различных газов совпадут, если они превратятся (воображаемо!) в идеальные газы.
Исторически сложилось так, что в течение многих десятилетий в качестве единицы давления использовалась атмосфера (атм.), причем 1 атм равна 1,01325105 Па. Нетрудно понять, что в стандартном состоянии газ должен находиться именно при этом давлении.
Хотя в последующие годы система единиц изменилась, давление идеального газа в стандартном состоянии осталось прежним, т.е. равным 1 атм.
Определение стандартного термодинамического состояния для газов таково:
Стандартное термодинамическое состояние газа при данной температуре представляет собой воображаемое состояние в виде идеального газа при давлении 1,01325105 Па.
Рассмотрим процесс перехода газа из стандартного состояния в заданное состояние, которому соответствует летучесть f.
Будем придерживаться следующего обязательного условия:
Все величины, относящиеся к стандартному состоянию или отсчитываемые от него, обозначаются символом о, который ставится вверху справа от определяемой величины.
По этой причине в стандартном состоянии давление и равная ему летучесть будут обозначаться следующим образом: fo = Po =1.01325105 Па.
Первая стадия перехода из стандартного состояния в заданное состояние газа включает расширение газа. Поскольку в стандартном состоянии он наделен свойствами идеального газа, то расширение его (не следует забывать, что речь идет об изотермической функции) должно происходить по изотерме идеального газа до очень маленького давления P* или летучести f*. Изменение энергии Гиббса на этой стадии равно
![]() .
.
При очень малых давлениях свойства реального газа фактически совпадают со свойствами идеального газа. Поэтому различие между изотермами идеального газа и изотермами реального газа в этих условиях отсутствуют. В связи с этим переход с изотермы идеального газа на изотерму реального газа не вызовет никаких изменений в системе. Следовательно, для второго этапа процесса изменение энергии Гиббса окажется равным нулю.
Третий этап представляет собой сжатие по изотерме реального газа от летучести f* до летучести в заданном состоянии f. Изменение энергии Гиббса на этом этапе равно
![]() .
.
Общее изменение энергии Гиббса в результате всех этапов равно
![]() .
.
Так как Po =fo и f*P*, то окончательно имеем
![]() (6 - 37)
(6 - 37)
или
![]() .
(6 - 38)
.
(6 - 38)
6 - 9. Стандартное термодинамическое состояние для конденсированных систем
В качестве стандартного термодинамического состояния при заданной температуре для жидких систем принимается состояние находящейся в равновесии с паром чистой жидкости, если ее агрегатное состояние наиболее устойчиво при этой температуре.
Приведем пример, иллюстрирующий это условие.
При температуре 298 К чистая вода, над которой имеется водяной пар, находится в стандартном состоянии. Воду можно осторожно охладить до температуры, намного меньшей температуры ее кристаллизации. В частности, экспериментально определено равновесное давление насыщенного пара над переохлажденной водой при температуре 257 К или -16оС. Однако при этой температуре вода, даже находящаяся в равновесии с водяным паром, не находится в стандартном состоянии, так как переохлажденная вода представляет собой неустойчивую систему. Достаточно внести в переохлажденную воду мельчайший кристаллик льда, чтобы немедленно началась ее кристаллизация.
Условие равновесия жидкости с паром в стандартном состоянии не означает, что давление пара над ней должно быть столь значительным, чтобы его можно было легко измерить. Например, при 240 К давление пара над чистой ртутью составляет всего лишь 10-3 Па. Эта величина очень мала, и пары ртути можно обнаружить только с помощью специальных средств. Однако возможность существования пара над чистой жидкостью уже является вполне достаточным условием для стандартного состояния.
Пар, находящийся в равновесии с жидкостью в стандартном состоянии, также находится в стандартном состоянии.
Это свойство отличает пар от газа.
Газом принято считать газообразное состояние вещества при отсутствии его равновесия с жидкостью, а паром - газообразное состояние вещества, находящегося в равновесии с жидкостью. Для каждого из них существует свое стандартное состояние.
Если пар имеет сравнительно низкое давление в стандартном состоянии и, следовательно, по свойствам приближается к идеальному газу, то его характеризуют стандартным давлением Ро. В других случаях используют стандартную летучесть fo.
Стандартным термодинамическим состоянием твердого вещества при заданной температуре является устойчивое состояние чистого кристаллического вещества при этой температуре.
Стеклообразное состояние, в котором вещество не имеет четкой кристаллической структуры, даже для чистых веществ не может рассматриваться как стандартное. Стеклование является результатом быстрого нарастания вязкости жидкости при охлаждении, в результате чего затрудняется образование кристаллической решетки. Иногда это состояние называют «замороженным неустойчивым состоянием».
6 - 10. Химические потенциалы веществ в реальных системах
Для того, чтобы найти зависимость химического потенциала вещества в газовой фазе от его летучести, проведем следующий воображаемый процесс.
Из системы, в которой газ i находится в стандартном состоянии и его химический потенциал равен io , перенесем его бесконечно малое число молей dni в реальную систему, в которой летучесть газа равна fi, а химический потенциал равен i.
Этот процесс сопровождается изменением энергии Гиббса
![]() ,
,
![]() .
(6 - 39)
.
(6 - 39)
С другой стороны, изменение летучести от fo до fi приводит к изменению энергии Гиббса для 1 моль
![]() ,
,
а для dni молей изменение энергии Гиббса составляет
![]() .
(6 - 40)
.
(6 - 40)
Приравнивая правые части равенств (6 - 39) и (6 - 40), получим
![]() .
.
Включая стандартную летучесть в общую константу, окончательно имеем
![]() (6 - 41)
(6 - 41)
Уравнение (6 - 41) отражает зависимость химического потенциала от летучести в реальных газовых смесях.
Обратимся к реальным растворам в конденсированных системах.
Пусть имеются две системы: конденсированная система в виде вещества i в стандартном состоянии и реальный раствор, содержащий это вещество.
Совершим следующий воображаемый процесс.
Перенесем бесконечно малое число молей dni вещества i из первой системы во вторую и найдем изменение энергии Гиббса на всех стадиях этого процесса.
Квазистатический переход из чистой жидкости (или твердого вещества) в пар не приводит к изменению энергии Гиббса. Переход dni моль из пара над чистым веществом с летучестью fio в пар над раствором с летучестью fi приводит к изменению энергии Гиббса
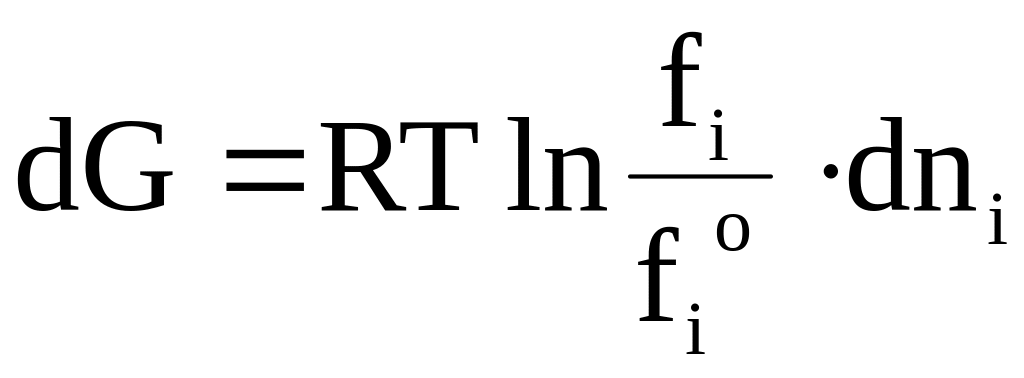 .
(6 - 42)
.
(6 - 42)
Последующий квазистатический переход из пара в жидкость не приводит к изменению энергии Гиббса. Таким образом, изменение энергии Гиббса для всего процесса определяется уравнением (6 - 42).
Если химический потенциал вещества i в стандартном состоянии равен io, а химический потенциал в растворе равен i, то изменение энергии Гиббса при переносе dni молей вещества из одной системы в другую можно представить следующим образом:
. (6 - 43)
Из равенства правых частей уравнений (6 - 42) и (6 ‑ 43) следует
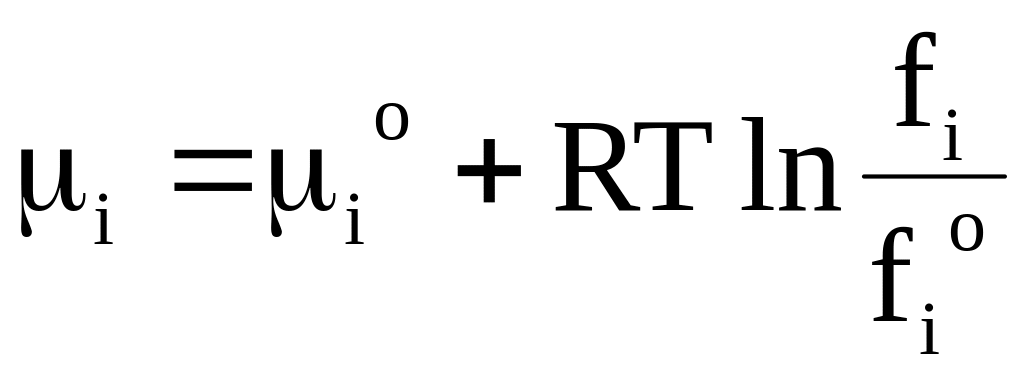 .
(6 - 44)
.
(6 - 44)
Уравнение (6 - 44), показывающее зависимость химического потенциала от состава конденсированной системы, имеет принципиальное отличие от подобного уравнения для газовых систем.
В газовых системах стандартная летучесть одинакова для всех газов, а для конденсированных систем стандартная летучесть является специфической величиной каждого вещества. Ее не включают в общую константу.
Отношение летучести вещества над его раствором к летучести в стандартном состоянии называется термодинамической активностью ai вещества в растворе
 (6 - 45)
(6 - 45)
В идеальных растворах термодинамическая активность совпадает с молярной долей вещества и выражение (6 - 45) можно рассматривать как обобщение закона Рауля для реальных систем.
Нетрудно заметить, что
в стандартном состоянии термодинамическая активность вещества равна 1.
Введение термодинамической активности упрощает запись основного выражения для химического потенциала, придавая ему форму:
![]() (6 - 46)
(6 - 46)