

Gosudarstvennaya_farmakopeya_RB
.pdfрежиме предел обнаружения значительно ниже. При использовании внутреннего
или внешнего стандартов (например, дейтерированные стандарты) могут быть проведены количественные или полуколичественные измерения. При
использовании времяпролётных анализаторов такие измерения невозможны.
Двойной фрагментометрический масс-спектрометрический режим
(множественный мониторинг реакций, multiple reaction monitoring (MRM)).
Включает специфическую мономолекулярную или бимолекулярная реакцию разрушения выбранного иона-предшественника, характерного для анализируемого вещества. Селективность и высокая специфичность данного режима получения
сигнала обеспечивают превосходную чувствительность и делают его наиболее подходящим для количественных определений с использованием подходящих
внутренних стандартов (например, дейтерированных стандартов). Данный вид анализа может быть проведен только при использовании приборов, снабжённых
тремя соединёнными квадруполями, ионными ловушками или циклотроннорезонансными анализаторами.
ГРАДУИРОВКА
Градуировка позволяет установить соответствие между величиной m/z и
детектируемым сигналом. Как правило, она проводится с использованием вещества
сравнения. Градуировка может быть внешней (данные находятся в другом файле)
или внутренней (вещество (вещества) сравнения смешивается с исследуемым веществом и результаты вносятся в файл с данными анализа). Число ионов или
точек, необходимое для надёжной градуировки, зависит от типа анализатора и
требуемой точности измерений, например, в случае магнитного анализатора, где отношение m/z экспоненциально изменяется при изменении величины магнитного поля необходимо брать настолько много точек, насколько это возможно.
ОБНАРУЖЕНИЕ СИГНАЛА И ОБРАБОТКА ДАННЫХ
Ионы, разделённые анализатором, преобразуются в электрические сигналы
такими детектирующими системами, как фотоумножитель или электронный
умножитель. Затем данные сигналы усиливаются и превращаются в цифровые сигналы, которые используются при обработке данных и позволяют проводить
различные операции: градуировку, получение спектров, автоматические
количественные расчёты, архивирование данных, создание или использование библиотек масс-спектров. Различные физические параметры, требуемые для работы прибора, в целом контролируются компьютером
2.2.44. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ОБЩЕГО ОРГАНИЧЕСКОГО УГЛЕРОДА В ВОДЕ ДЛЯ ФАРМАЦЕВТИЧЕСКОГО ПРИМЕНЕНИЯ
Определение содержания общего органического углерода (ООУ) является непрямым методом определения содержания сумм органических веществ в воде
для фармацевтического применения. Определение содержания ООУ может также использоваться при контроле выполнения различных операций при производстве лекарственных средств.
Поскольку для определения содержания ООУ могут применяться различные
методики, в данной статье приведены не описания методик, а их квалификация и интерпретация результатов в предельных испытаниях. Испытания стандартного
раствора проводят через определенные интервалы времени, в зависимости от частоты измерений; раствор готовят с использованием легкоокисялющейся
субстанции (например, сахароза) с такой концентрацией, чтобы отклик прибора соответствовал измеряемому пределу содержания ООУ. Пригодность системы проверяют с использованием трудноокисляющейся субстанции (например, 1,4- бензохинон).
Разные типы приборов для определения ООУ в воде для фармацевтического применения, как правило, предназначены для полного окисления органических
молекул в образце воды до углерода диоксида с последующим измерением его количества, используемого затем для расчета концентрации углерода в воде.
Используемый прибор в процессе работы может определять органический и неорганический углерод (неорганический углерод присутствует в виде карбонатов).
Это может обеспечиваться путем определения количества неорганического углерода и вычитанием его из количества общего углерода или удалением неорганического углерода из образца при помощи продувания перед окислением. Органические молекулы могут удаляться с испытуемого образца в процессе продувания, но часть углерода, связанного с ими, в воде для фармацевтического
применения, незначительна.
Прибор. Используют отградуированный прибор, установленный в режим “он-
лайн” или автономно. Пригодность системы проверяют, как описано ниже, через
определенный промежуток времени. Прибор должен иметь разрешающую способность определения углерода - 0,05 мг/л или менее, соответственно паспорту тзготовителя прибора.
Вода для определения содержания ООУ. Используют воду высокоочищенную, соответствующую требованиям:
-удельная электропроводность: не более 1,0 мкСм·см-1 при температуре 25оС;
-содержание общего органического углерода: не более 0,1 мг/л.
Взависимости от типа используемого прибора, критическим параметром может быть также содержание в воде тяжелых металлов или меди, что должно
быть указано в инструкции изготовителя прибора.
Подготовка посуды. Используют посуду, тщательно вымотую методом,
позволяющим удалить органические вещества. Для последнего промывания
используют воду для определения содержания ООУ.
Стандартный раствор. Сахарозу Р, предварительно высушенную при
температуре 105оС в течение 3 ч, растворяют в воде для определения ООУ,
получая раствор, содержащий 1,19 мг/л сахарозы (0,50 сг/л углерода).
Испытуемый раствор. Испытуемую воду собирают, исключая минимальное воздушное пространство, в плотноукупоренный контейнер, используя приспособление для предотвращения загрязнения. Испытание проводят сразу с
целью устранения загрязнения воды от контейнера и его закрывающегося
ответстия.
Раствор для проверки пригодности системы. Готовят раствор 0,75 мг/л
1,4-бензохинона Р в воде для определения ООУ (0,50 мг/л углерода).

Контрольная вода для определения содержания ООУ. Используют воду
для определения содержания ООУ, полученную одновременно с водой для приготовления стандартного раствора для проверки пригодности системы.
Контрольный раствор. Кроме контрольной воды для определения
содержания ООУ, готовят подходящий контролный раствор или другие растворы,
необходимые для восстановления базовой линии или корригирования градуировки в соответствии с инструкцией изготовителя прибора; устанавливают ноль прибора с использованием контрольных растворов.
Проверка пригодности системы. Проводят испытания указанных растворов и
записывают отклики прибора; эффективность откликов, в процентах, рассчитывают по формуле:
rSS - rW ×100, rS - rW
где:
rW – отклик прибора, соответствующий воде для определения содержания ООУ; rS – отклик прибора, соответствующий стандартному раствору;
rSS – отклик прибора, соответствующий раствору для проверки пригодности
системы.
Система считается пригодной, если эффективность отклика прибора составляет не менее 85% и не более 115% от теоретического отклика.
Методика. Записывают отклик ( rU ) для испытуемого раствора. Испытуемый раствор выдерживает испытание, если значение rU не превышает значение rS - rW .
Данная методика может быть выполнена в режиме «он-лайн» на приборе,
который соответствующим образом отградуирован и соответствует требованиям пригодности системы. Прибор должен быть установлен таким образом, чтобы
обеспечить показания откликов, которые соответствуют испытуемой воде.
2.2.45. СВЕРХКРИТИЧЕСКАЯ ФЛЮИДНАЯ ХРОМАТОГРАФИЯ
Сверхкритическая флюидная хроматография (СФХ, SFC) - это метод хроматографического разделения, в котором подвижная фаза является флюидом,
находящимся в сверхкритическом или субкритическом состоянии. Неподвижная
фаза, содержащаяся в колонке, состоит либо из тонко измельчённых твёрдых частиц, таких как силикагель или пористый графит; химически модифицированная
неподвижная фаза, такая же, как и в жидкостной хроматографии, или, в случае
капиллярных колонок, плёнка жидкости, равномерно нанесённая на стенки колонки. СФХ основана на механизмах адсорбции или распределения.
АППАРАТУРА
Прибор обычно состоит из охлаждённой насосной системы, инжектора, хроматографической колонки, находящейся в термостатируемой печи, детектора, регулятора давления и регистрирующего устройства (интегратор или самописец).
Насосная система.
Насосная система необходима для поддержания постоянной скорости
подвижной фазы. Колебания давления должны быть сведены к минимуму, например, путём пропускания сжимаемой жидкости через устройство, подавляющее
пульсацию. Насосно-компрессорная система и соединительные линии устойчивы к давлениям, создаваемым насосом.
Точная доставка подвижной фазы при постоянных либо изменяющихся по определённой программе условиях осуществляется системами, контролируемыми микропроцессором. В случае градиентного элюирования могут быть использованы насосные системы, доставляющие растворитель (растворители) из нескольких
резервуаров, смешивание растворителей может быть проведено как со стороны насоса (насосов), имеющей низкое давление, так и со стороны, имеющей высокое
давление.
Инжекторы
Ввод пробы может быть осуществлён непосредственно в колонку с помощью клапана.
Неподвижные фазы
Неподвижные фазы находятся в колонках, которые были описаны в главах,
посвящённых Жидкостной хроматографии (2.2.29) (набивные колонки) и Газовой хроматографии (2.2.28) (капиллярные колонки). Максимальный внутренний
диаметр (Ø) капиллярной колонки равен 100 мкм.
Подвижные фазы
Обычно подвижной фазой является диоксид углерода, который может
содержать полярный модификатор, такой как метанол, 2-пропанол или
ацетонитрил. Состав, давление (плотность), температура и скорость используемой подвижной фазы могут быть как постоянными в течение всего хроматографического процесса (изократическое, изотермическое элюирование, элюирование при постоянной плотности), так и изменяться по определённой программе (градиентное элюирование модификатора, давления (плотности), температуры или скорости потока).
Детекторы
Наиболее часто используемыми детекторами являются спектрофотометры, работающие в ультрафиолетовой и видимой областях (UV/Vis) и пламенно-
ионизационные детекторы. Кроме того, могут применяться детекторы, работа
которых основана на измерении рассеяния света, ИК-спектрофотометры, катарометры или другие специальные детекторы.
МЕТОД
Исследуемый раствор (растворы) и раствор (растворы) сравнения готовят так, как описано в методике. Растворы не должны содержать твёрдых частиц.
Критерии для оценки пригодности системы были описаны в главе
Хроматографические методы разделения (2.2.46). В данной главе были также
приведены уровни, до которых можно регулировать параметры хроматографической системы, для того, чтобы она оставалась пригодной.
2.2.46. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ РАЗДЕЛЕНИЯ
Хроматографическими называют многостадийные методы разделения, в
которых компоненты образца распределяются между двумя фазами, одна из
которых является неподвижной, а другая – подвижной. Неподвижная фаза может
быть твёрдым веществом, жидкостью, нанесённой на твёрдый носитель, либо гелем. Неподвижная фаза помещается в колонку, распределяется в виде тонкого
слоя, плёнки и т.д. Подвижная фаза может быть газом, жидкостью или сверхкритическим газом (флюидом). Разделение основано на адсорбции,
распределении, ионном обмене и т.д., либо на различиях в физико-химических свойствах молекул, таких как размер, масса, объём и т.д.
Данная глава содержит определения и расчёты общих параметров и применимых ко всем хроматографическим методам требований для пригодности
системы. Принципы разделения, аппаратура и методики приводятся в следующих общих статьях:
Бумажная хроматография (2.2.26) Тонкослойная хроматография (2.2.27)
Газовая хроматография (2.2.28) Жидкостная хроматография (2.2.29)
Эксклюзионная хроматография (2.2.30)
Сверхкритическая флюидная хроматография (2.2.45) ОПРЕДЕЛЕНИЯ
Для расчёта пределов в монографиях использованы следующие
определения.
При использовании некоторого оборудования ряд параметров, например отношение сигнал/шум, может быть рассчитано с помощью программного
обеспечения, поставляемого производителем. На пользователе лежит
ответственность за соответствие способов расчётов, используемых в программном обеспечении, требованиям Фармакопеи. В противном случае должны быть сделаны соответствующие исправления.
Хроматограмма
Хроматограмма представляет собой графическое или иное представление сигнала детектора на концентрацию веществ в элюате или другой количественной
величины, используемой для измерения концентрации веществ в элюате,
относительно времени, объёма или расстояния. Идеализированные хроматограммы представлены сочетанием гауссовских пиков и базовой линии
(базовой линии).
ПАРАМЕТРЫ УДЕРЖИВАНИЯ Время удерживания и удерживаемый объём
Измерения удерживания в элюентной хроматографии могут быть представлены временем удерживания (tR), определённом непосредственно по
положению максимума пика на хроматограмме. Из времени удерживания может быть рассчитан удерживаемый объём (VR).
VR = tR v
tR – время удерживания или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика, соответствующего компоненту,

v – объёмная скорость подвижной фазы.
Концентрационный коэффициент распределения
Концентрационный коэффициент распределения (Dm) (также известный как коэффициент ёмкости k’ или фактор удерживания k) определяется как:
Dm = |
количество × вещества × в × неподвижной × фазе |
= KC |
VS |
|
количество × вещества × в × подвижной × фазе |
VM |
|||
|
|
KC – коэффициент распределения,
VS – объём неподвижной фазы,
VM – объём подвижной фазы.
Значение Dm компонента может быть определено из хроматограммы при использовании выражения:
Dm = tR t- tM
M
tR – время удерживания (или объём) или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика,
соответствующего компоненту,
tM – «мёртвое» время (или объём): время (или объём) или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из
максимума пика, соответствующего неудерживаемому компоненту.
Коэффициент распределения
Характеристика элюирования в эксклюзионной хроматографии может быть
представлена коэффициентом распределения (K0), который рассчитывают с
помощью выражения:
K0 = ttR -tt0
t - 0
tR – время удерживания (или объём) или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика,
соответствующего компоненту,
t0 – «мёртвое» время (или объём): время (или объём) или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика, соответствующего неудерживаемому компоненту,
tt – время удерживания (или объём) или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика,
соответствующего компоненту, который полностью проникает в поры неподвижной фазы.
Фактор удерживания (подвижности)
Фактор удерживания (подвижности RF ), используемый в плоскостной хроматографии, представляет собой отношение расстояния от точки нанесения пробы до центра пятна и расстояния, пройденного фронтом растворителя от точки нанесения пробы.

RF = ba b – расстояние, пройденное веществом,
a – расстояние, пройденное фронтом растворителя.
ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
Пик может быть охарактеризован площадью пика (A) или высотой пика (h) и
шириной пика на половине высоты (wh) или высотой пика (h) и шириной пика между точками перегиба (wi). Для гауссовских пиков (Рисунок 2.2.46.-1) справедливо соотношение:
wh = 1,18wi
Рисунок 2.2.46.-1.
Фактор асимметрии
Фактор асимметрии пика (As) (Рисунок 2.2.46.-2) рассчитывается из
выражения:
As = w0,05
2d w0,05 – ширина пика на одной двадцатой высоты,
d – расстояние между перпендикуляром, опущенным из максимума пика и передним краем пика на одной двадцатой высоты.
Если фактор ассиметрии равен 1,0 – это означает полную (идеальную) симметрию.
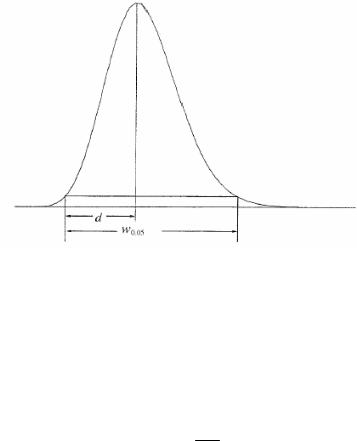
Рисунок 2.2.46.-2.
Эффективность колонки и число теоретических тарелок
Эффективность колонки, в зависимости от метода, может быть рассчитана из данных, полученных как при изотермическом, изократическом режимах, так и при
режиме постоянной плотности, как число теоретических тарелок (n) из следующего
выражения, в котором величины tR и wh должны быть выражены в одинаковых единицах (время, объём или расстояние).
N 5,54æ tR ö2
= ççè wh ÷÷ø
tR – время удерживания (или объём) или расстояние, измеренное по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика, соответствующего компоненту,
wh – ширина пика на половине высоты.
Число теоретических тарелок зависит от компонента, колонки и времени
удерживания.
ПАРАМЕТРЫ РАЗДЕЛЕНИЯ Разрешение
Разрешение (Rs) близких по высоте пиков двух компонентов может быть
рассчитано из выражения:
Rs = |
1,18(tR2 − tR1 ) |
|
wh1 + wh2 |
||
|
tR2 > tR1
tR1 и tR2 – времена удерживания или расстояния, измеренное по базовой линии от точки ввода пробы до перпендикуляров, опущенных из максимумов двух соседних
пиков,
wh1 и wh2 – ширина пиков на половине высоты.
Разрешение более 1,5 соответствует разделению пиков до базовой линии.

Выражение, приведенное выше, не может быть использовано в случае, когда
пики сильно различаются по высоте.
В количественной плоскостной хроматографии вместо времён удерживания
используются пройденные расстояния, и разрешение может быть рассчитано с использованием выражения:
RS = |
1,18a(RF2 − RF1 ) |
|
wh1 + wh2 |
||
|
RF1 и RF2 – отношения расстояний от точки нанесения пробы до центров пятен и
расстояния, прошедшего фронтом растворителя от точки нанесения пробы (фактор подвижности),
wh1 и wh2 – ширина пиков на половине высоты,
a – расстояние, прошедшее фронтом растворителя.
Коэффициент разделения пиков
Коэффициент разделения неполностью разделённых пиков (p/v-параметр)
может быть использован в качестве требования пригодности хроматографической системы при выполнении испытания на родственные соединения в случае неполного отделения примеси от анализируемого вещества (аналита) (Рисунок
2.2.46.-3).
p / v = H p H v
Hp – высота пика примеси относительно экстраполированной базовой линии,
Hv – расстояние между экстраполированной базовой линией и нижней точкой
кривой, разделяющей пики примеси и аналита.
Рисунок 2.2.46.-3.
Относительное удерживание
Относительное удерживание (r) рассчитывается из выражения:

r= tR2 − tM tR1 − tM
tR2 – время удерживания интересующего пика,
tR1 – время удерживания пика сравнения (обычно пик, соответствующий
исследуемому веществу),
tM – «мёртвое» время (или объём): время (или объём) или расстояние, измеренное
по базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума пика, соответствующего неудерживаемому компоненту.
В плоскостной хроматографии вместо tR2 и tR1 используются факторы подвижности
RF2 и RF1.
ВОСПРОИЗВОДИМОСТЬ (ПРЕЦИЗИОННОСТЬ) КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ
Отношение сигнал/шум
Отношение сигнал/шум (S/N) влияет на воспроизводимость (прецизионность) количественного определения и рассчитывается из уравнения:
NS = 2hH
H – высота пика (Рисунок 2.2.46.-4), соответствующая рассматриваемому
компоненту, на хроматограмме, полученной при использовании рекомендованного раствора сравнения, измеренная от максимума пика до экстраполированной
базовой линии для сигнала, величина которого эквивалентна двадцатикратному
превышению величины ширины пика на половине высоты,
h – размах для фонового шума (уровень шума) на хроматограмме, полученной при введении или нанесении холостой пробы, величина которого эквивалентна двадцатикратному превышению щирины пика на половине высоты для пика на хроматограмме, полученной при использовании рекомендованного раствора сравнения и, если возможно, расположенного на одном и том же расстоянии от
места возможного обнаружения пика.
Рисунок 2.2.46.-4.