

Gosudarstvennaya_farmakopeya_RB
.pdf2.4.23. СТЕРИНЫ В ЖИРНЫХ МАСЛАХ
ОТДЕЛЕНИЕ СТЕРИНОВОЙ ФРАКЦИИ
Получают неомыляемые вещества жирного масла и затем отделяют стериновую фракцию методом тонкослойной хроматографии (2.2.27), используя пластинки,
покрытые тонким слоем силикагеля G P толщиной от 0,3 мм до 0,5 мм. Испытуемый раствор (а). В колбу вместимостью 150 мл, снабженную
обратным холодильником, помещают раствор 2 г/л бетулина Р в метиленхлориде Р в таком объеме, чтобы количество бетулина соответствовало около 10% содержания стеринов в образце, взятом для
определения (т.е. в случае оливкового масла прибавляют 500 мкл, в случае других растительных масел 1500 мкл раствора бетулина). Выпаривают до
сухого остатка в токе азота Р. Если в частной статье определяется только содержание индивидуальных стеринов в процентах к стериновой фракции, раствор бетулина можно не прибавлять.
В колбу помещают 5,00 г испытуемого вещества. Прибавляют 50 мл 2 М
раствора калия гидроксида спиртового Р и нагревают на водяной бане в течение 1 ч, часто перемешивая содержимое колбы круговыми движениями. Охлаждают до температуры ниже 25оС и количественно
переносят содержимое колбы в делительную воронку, содержащую 100 мл воды Р. Осторожно встряхивают с тремя порциями эфира, свободного от пероксидов Р, по 100 мл каждая. Эфирные слои собирают в другую
делительную воронку, содержащую 40 мл воды Р, слегка встряхивают в
течение нескольких минут, оставляют до расслоения и отбрасывают водный слой. Эфирный слой встряхивают с несколькими порциями воды Р, по 40 мл каждая, до тех пор, пока водный слой не перестанет давать щелочную реакцию по фенолфталеину. Эфирный слой количественно
переносят в колбу, высушенную до постоянной массы, промывая
делительную воронку эфиром, свободным от пероксидов Р. Эфир выпаривают с необходимыми предосторожностями, к остатку прибавляют 6 мл ацетона Р. Осторожно удаляют растворитель в токе азота Р. Высушивают до постоянной массы при температуре от 100°С до 105°С,
охлаждают в эксикаторе и взвешивают. Остаток растворяют в минимальном
объеме метиленхлорида Р.
Испытуемый раствор (b). В колбу вместимостью 150 мл, снабженную обратным холодильником, помещают 5,00 г рапсового масла Р и далее поступают, как указано в приготовлении испытуемого раствора (а), начиная
со слов «Прибавляют 50 мл 2 М раствора калия гидроксида спиртового Р».
Испытуемый раствор (с). В колбу вместимостью 150 мл, снабженную
обратным холодильником, помещают 5,00 г подсолнечного масла Р и далее поступают, как указано в приготовлении испытуемого раствора (а), начиная со
слов «Прибавляют 50 мл 2 М раствора калия гидроксида спиртового Р». Раствор сравнения. 25 мг холестерина Р и 10 мг бетулина Р растворяют
в 1 мл метиленхлорида Р.
Для каждого испытуемого раствора используют отдельную пластинку. На каждую из пластинок наносят полосой размером (20x3) мм 20 мкл раствора
сравнения и полосой размером (40x3) мм 400 мкл испытуемого раствора (а) или испытуемого раствора (b), или испытуемого раствора (с), соответственно.
Пластинки помещают в камеру со смесью растворителей эфир Р - гексан
Р (35:65). Когда фронт растворителей пройдет 18 см от линии старта, пластинки вынимают из камеры, высушивают в токе азота Р, опрыскивают
раствором 2 г/л дихлорфлуоресцеина Р в этаноле Р и просматривают в УФ–
свете при длине волны 254 нм. На хроматограмме раствора сравнения
обнаруживаются полосы, соответствующие холестерину и бетулину. На
хроматограммах испытуемых растворов обнаруживаются полосы с
близкими значениями Rf, соответствующие стеринам. С хроматограммы каждого испытуемого раствора снимают область покрытия, на которой
расположены полосы стеринов, а также дополнительно зоны на 2-3 мм выше
и ниже видимых зон раствора сравнения и помещают раздельно в три колбы вместимостью 50 мл каждая. В каждую колбу прибавляют по 15 мл теплого метиленхлорида Р и встряхивают. Каждый раствор фильтруют
через стеклянный фильтр (40) или подходящую фильтровальную бумагу и промывают каждый фильтр тремя порциями метиленхлорида Р, по 15 мл
каждая. Объединенные фильтраты и смывы помещают в три колбы, высушенные до постоянной массы, выпаривают до сухого остатка в токе
азота Р и взвешивают.
ОПРЕДЕЛЕНИЕ СТЕРИНОВ Определение стеринов проводят методом газовой хроматографии
(2.2.28).
Все операции проводят, защищая растворы и реактивы от влаги, растворы готовят непосредственно перед применением.
Испытуемый раствор. К выделенным из испытуемого вещества методом
тонкослойной хроматографии стеринам прибавляют свежеприготовленную смесь: хлортриметилсилан - гексаметилдисилазан Р - пиридин безводный
Р (1:3:9). (0,02 мл смеси на миллиграмм остатка). Осторожно встряхивают до
полного растворения остатка и оставляют в эксикаторе над фосфора (v) оксидом Р на 30 мин. При необходимости центрифугируют и используют надосадочную жидкость.
Раствор сравнения (а). К 9 частям стеринов, выделенных из рапсового масла
Рметодом тонкослойной хроматографии, прибавляют 1 часть холестерина
Р. К полученной смеси прибавляют свежеприготовленную смесь:
хлортриметилсилан - гексаметилдисилазан Р - пиридин безводный Р
(1:3:9) (0,02 мл смеси на миллиграмм смеси стеринов). Осторожно
встряхивают до полного растворения остатка и оставляют в эксикаторе над фосфора (V) оксидом Р на 30 мин. При необходимости центрифугируют и используют надосадочную жидкость.
Раствор сравнения (b). К стеринам, выделенным из подсолнечного масла Р
методом тонкослойной хроматографии, прибавляют свежеприготовленную
смесь: хлортриметилсилана Р - гексаметилдисилазан - пиридин безводный Р (1:3:9) (0,02 мл смеси на миллиграмм смеси стеринов).
Осторожно встряхивают до полного растворения остатка и оставляют в эксикаторе над фосфора (V) оксидом Р на 30 мин. При необходимости
центрифугируют и используют надосадочную жидкость.
Хроматографируют на газовом хроматографе с пламенно-ионизационным детектором в следующих условиях:
-кварцевая колонка длиной от 20 м до 30 м и внутренним диаметром от 0,25
мм до 0,32 мм, покрытая поли[метил(95)фенил(5)]силоксаном Р или поли[метил(94)фенил(5)винил(1)]силоксаном Р с толщиной слоя 0,5 мкм,
-газ-носитель – водород для хроматографии Р (линейная скорость от 30
см/с до 50 см/с) или гелий для хроматографии Р (линейная скорость от 20 см/с до 35 см/с). Линейную скорость измеряют следующим способом: в

условиях определения стеринов вводят от 1 мкл до 3 мкл метана или
пропана. Измеряют время в секундах, необходимое для прохождения газа через колонку от момента введения до появления пика (tm).
Линейную скорость вычисляют по формуле L/tm, где L - длина колонки в сантиметрах;
-деление потока – 1:50 или 1:100;
-температура колонки – 260°С;
-температура инжектора – 280°С;
-температура детектора – 290°С.
Хроматографируют в указанных условиях по 1 мкл каждого раствора.
На хроматограмме раствора сравнения (а) обнаруживается четыре основных пика, соответствующих холестерину, брассикастерину,
кампестерину и β-ситостерину; на хроматограмме раствора сравнения (b) обнаруживается четыре основных пика, соответствующих кампестерину, стигмастерину, β-ситостерину и 7-сигмастенолу. Времена удерживания
стеринов относительно β-ситостерина даны в Табл. 2.4.23.-1.
Пик внутреннего стандарта (бетулин) должен быть четко отделен от пиков определяемых стеринов.
Идентифицируют пики, обнаруженные на хроматограмме испытуемого
раствора, и вычисляют процентное содержание каждого стерина в стериновой фракции испытуемого вещества по формуле:
С = SA ×100
где:
С ― процентное содержание стеарина; А ― площадь пика, соответствующего определяемому стерину;
S ― сумма площадей всех пиков, соответствующих компонентам, указанным в Табл. 2.4.23.-1.
Если это указано в частной статье, вычисляют количество каждого стерина
вмиллиграммах, содержащееся в 100 г испытуемого вещества, по формуле:
С= A × mS ×100
AS × m
где:
С ―количество стеарина в миллиграммах;
А ― площадь пика, соответствующего определяемому компоненту;
As ― площадь пика, соответствующего бетулину;
m ― масса навески образца испытуемого вещества, в граммах; ms ― масса прибавленного бетулина Р, в миллиграммах.
Таблица 2.4.23.-1
Времена удерживания стеринов по отношению к времени удерживания β-ситостерина для двух различных колонок
|
Поли[метил(9 5 ) - |
Поли[метил(94)-фенил(5) |
|
фенил(5)]силоксан |
вннил(1)]-силоксан |
|
|
|
Холестерин |
0.63 |
0.67 |
Брассикастерин |
0.71 |
0.73 |
24-Метиленхолестерин |
0.80 |
0.82 |
Кампестерин |
0.81 |
0.83 |
Кампестанол |
0.82 |
0.85 |
Стигмастерин |
0.87 |
0.88 |
∆-Кампестерин |
0.92 |
0.93 |
∆5,23-Стигмастадиенол |
0.95 |
0.95 |
Клеростерин |
0.96 |
0.96 |
β-Ситостерин |
1 |
1 |
Ситостанол |
1.02 |
1.02 |
∆5-Авенастерин |
1.03 |
1.03 |
∆5,24-Стигмастадиенол |
1.08 |
1.08 |
∆7-Стигмастенол (1) |
1.12 |
1.12 |
∆7-Авенастерин |
1.16 |
1.16 |
Бетулин |
1.4 |
1.6 |
(1) ∆7-Стигмастенол в литературе может упоминаться как ∆7-Стигмастерин
2.4.24. ИДЕНТИФИКАЦИЯ ОСТАТОЧНЫХ РАСТВОРИТЕЛЕЙ И ИХ КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Методики испытаний, приведенные в этой общей статье, могут быть
использованы:
1)для идентификации основной массы остаточных растворителей классов 1 и 2 в субстанциях, вспомогательных веществах и готовых лекарственных средствах, если остаточные растворители неизвестны;
2)для определения предельного содержания растворителей классов 1 и 2, если они присутствуют в субстанциях, вспомогательных веществах и готовых лекарственных средствах;
3)для количественного определения растворителей класса 2, если их
предельное содержание превышает 1000 ppm (0,1%), или, если необходимо, для количественного определения растворителей класса 3.
Остаточные растворители классов 1, 2 и 3 перечислены в статье 5.4.
«Остаточные количества органических растворителей».
Для приготовления испытуемых растворов ниже приведены три растворителя,
а также условия введения газовых проб при парофазной газовой хроматографии.
Хотя описаны две хроматографические системы, предпочтительнее использовать систему А, система В обычно используется для идентификации. Выбор методики
приготовления испытуемых растворов зависит от растворимости анализируемого вещества и, в первую очередь, от класса контролируемого остаточного
растворителя.
Формамид, 2-этоксиэтанол, 2-метоксиэтанол, этиленгликоль, N-
метилпирролидон и сульфолан следует определать другими валидированными методиками.
Если приведенную ниже методику испытаний применяют для количественного
определения остаточных растворителей в конкретном веществе, она должна быть валидирована.
МЕТОДИКА
Испытания проводят методом парофазной газовой хроматографии (2.2.28).
Пробоподготовка 1. Применяют при определении остаточных растворителей
в веществах, растворимых в воде.
Исходный раствор испытуемого образца (1). 0,250 г испытуемого образца растворяют в воде Р и доводят объем раствора этим же растворителем до 25,0 мл.
Пробоподготовка 2. Применяют при определении остаточных растворителей в веществах, нерастворимых в воде.
Исходный раствор испытуемого образца (2). 0,250 г испытуемого образца растворяют в N,N-диметилформамиде Р (ДМФ) и доводят объем раствора этим же растворителем до 25,0 мл.
Пробоподготовка 3. Применяют при контроле N,N-диметилацетамида и/или
N,N-диметилформамида, если известно или допускается, что один или оба
растворителя присутствуют в испытуемом образце.
Исходный раствор испытуемого образца (3). 0,250 г испытуемого образца растворяют в 1,3-диметил-2-диметил-2-имидазолидоне Р (ДМИ) и доводят объем раствора этим же растворителем до 25,0 мл.
Если ни одна из вышеприведенных методик пробоподготовки не подходит,
возможно использование другого растворителя и других подходящих условий парофазного анализа.
Раствор остаточного растворителя (а). 1,0 мл ФСО раствора остаточного растворителя класса 1 доводят водой Р до 100,0 мл. 1,0 мл полученного раствора доводят водой Р до 10,0 мл.
Раствор остаточных растворителей (b). Соответствующие количества остаточных растворителей класса 2 растворяют в диметилформамиде Р и доводят объем водой Р до 100,0 мл. Объем полученного раствора доводят водой Р до
концентрации, соответствующей 1/20 значений пределов концентраций, указанных в Табл.1 или Табл.2 статьи 5.4. «Остаточные количества органических
растворителей».
Раствор остаточных растворителей (с). 1,00 г растворителя или растворителей, присутствующих в испытуемом образце, растворяют в
диметилсульфоксиде Р или в воде Р и доводят объем раствора водой Р до
концентрации, соответствующей 1/20 значений пределов концентраций, указанных в Табл.1 или Табл.2 статьи 5.4. «Остаточные количества органических
растворителей».

Холостой раствор (контроль). Готовят аналогично раствору остаточных
растворителей (с), но без добавления остаточных растворителей (используют для проверки отсутствия мешающих пиков).
Испытуемый раствор. 5,0 мл исходного раствора испытуемого образца,
подготовленного в соответствии с методикой пробоподготовки, и 1,0 мл холостого раствора помещают во флакон.
Раствор сравнения (а) (класс 1). 1,0 мл раствора остаточного растворителя
(а) и 5,0 мл подходящего растворителя помещают во флакон.
Раствор сравнения (а1) (класс 1). 5,0 мл исходного раствора испытуемого образца, подготовленного в соответствии с методикой пробоподготовки, и 1,0 мл
раствора остаточного растворителя (а) помещают во флакон.
Раствор сравнения (b) (класс 2). 1,0 мл раствора остаточного растворителя
(b) и 5,0 мл подходящего растворителя помещают во флакон.
Раствор сравнения (с). 5,0 мл исходного раствора испытуемого образца, подготовленного в соответствии с методикой пробоподготовки, и 1,0 мл раствора
остаточного растворителя (с) помещают во флакон.
Раствор сравнения (d). 1,0 мл холостого раствора и 5,0 мл подходящего
растворителя помещают во флакон.
Флаконы плотно закрывают резиновыми пробками, покрытыми политетрафторэтиленом, и обжимают алюминиевыми колпачками. Встряхивают до получения однородного раствора.
Для проведения парофазного анализа могут быть использованы следующие
условия:
Параметры |
|
Методика пробоподготовки |
|
||
хроматографической |
1 |
|
2 |
|
3 |
системы |
|
|
|
|
|
Равновесная температура |
80 |
|
105 |
|
80 |
(оС) |
|
|
|
|
|
Время достижения |
60 |
|
45 |
|
45 |
равновесия (мин) |
|
|
|
|
|
Температура линии подачи |
85 |
|
110 |
|
105 |
газовой пробы (оС) |
|
|
|
|
|
Газ-носитель: азот для хроматографии Р или гелий для хроматографии Р под
соответствующим давлением
Время нахождения под |
30 |
30 |
30 |
давлением (с) |
|
|
|
Объем вводимой пробы |
1 |
1 |
1 |
(мл) |
|
|
|
Для проведения хроматографического анализа могут быть использованы следующие системы.
Система А:
- колонка кварцевая капиллярная 30 м х 0,32 мм или 30 м х 0,53 мм, покрытая
слоем поперечносшитого полимера с 6% полицианопропилфенилсилоксана и 94% полидиметилсилоксана толщиной 1,8 мкм или 3 мкм;
-газ-носитель: азот для хроматографии Р или гелий для хроматографии Р, с
делением потока 1:5 и линейной скоростью газа-носителя около 35 см/с;
-пламенно-ионизационный детектор (для хлорсодержащих остаточных растворителей класса 1 может быть также использован масс-
спектрометрический детектор или детектор электронного захвата);
-температура колонки : изотермический режим при 40оС в течение 20 минут с
последующим программированным повышением температуры со скоростью
10оС/мин до 240оС, затем изотермический режим при температуре 240оС в течение 20 минут;
-температура блока ввода проб - 140 оС;
-температура детектора -250 оС.
Если возможно мешающее влияние основного вещества (матрицы), используют
систему В.
Система В:
- колонка кварцевая капиллярная 30 м х 0,32 мм или 30 м х 0,53 мм, покрытая слоем макрогола 20 000 Р толщиной 0,25 мкм;
-газ-носитель: азот для хроматографии Р или гелий для хроматографии Р, с
делением потока 1:5 и линейной скоростью газа-носителя около 35 см/с;
-пламенно-ионизационный детектор (для хлорсодержащих остаточных растворителей класса 1 может быть также использован масс- спектрометрический детектор или детектор электронного захвата);
-температура колонки : изотермический режим при 50оС в течение 20 минут с
последующим программированным повышением температуры со скоростью 6оС/мин до 165оС, затем изотермический режим при температуре 165оС в
течение 20 минут;
-температура блока ввода проб -140 оС;
-температура детектора -250 оС.
Проверка пригодности хроматографической системы А
Хроматографическая система А считается пригодной, если выполняются следующие условия:
- вводят 1 мл равновесной газовой фазы раствора сравнения (а1) и
записывают хроматограмму в таких условиях, чтобы можно было измерить отношение сигнал/шум для 1,1,1-трихлорэтана; отношение сигнал/шум должно быть не менее 5.
Типичная хроматограмма представлена на Рис. 2.4.24.-1; - вводят 1 мл равновесной газовой фазы раствора сравнения (а) в колонку;
пики, соответствующие |
остаточным растворителям класса 1 должны |
обнаруживаться; |
|
- вводят 1 мл равновесной газовой фазы раствора сравнения (b) в колонку и записывают хроматограмму в таких условия, чтобы можно было определить степень
разделения между пиками ацетонитрила и метиленхлорида; хроматографическая система пригодна, если полученная хроматограмма имеет вид хроматограммы,
представленной на Рис. 2.4.24.-2, а степень разделения между пиками ацетонитрила и метиленхлорида - не менее 1,0.
Выполнение измерений в системе А:
- вводят 1 мл равновесной газовой фазы испытуемого раствора в колонку. Если на полученной хроматограмме не обнаруживаются пики, соответствующие
пикам остаточных растворителей на хроматограммах, полученных для растворов
сравнения (а) или (b), то анализируемый образец выдерживает испытания. Если какой-
либо пик на полученной хроматограмме испытуемого раствора соответствует одному из пиков остаточных растворителей на хроматограммах, полученных для растворов сравнения (а) или (b), то должна быть применена система В.
Проверка пригодности хроматографической системы В
Хроматографическая система В считается пригодной, если выполняются следующие условия:
-вводят 1 мл равновесной газовой фазы раствора сравнения (а1) в колонку и
записывают хроматограмму в таких условиях, чтобы можно было замерить
соотношение сигнал/шум для бензола; отношение сигнал/шум должно не менее 5 (типовая хроматограмма представлена на Рис.2.4.24.-3).
-вводят 1 мл равновесной газовой фазы раствора сравнения (а) в колонку;
пики, соответствующие |
остаточным растворителям класса 1 должны |
обнаруживаться; |
|
- вводят 1 мл равновесной газовой фазы раствора сравнения (б) в колонку и записывают хроматограмму в таких условия, чтобы можно было определить степень разделения между пиками ацетонитрила и трихлорэтилена; система пригодна, если полученная хроматограмма имеет вид хроматограмы, представленной на Рис. 2.4.24.-4, а степень разделения пиков ацетонитрила и трихлорэтилена - не менее
1,0.
Выполнение измерений в системе В
Вводят 1 мл равновесной газовой фазы испытуемого раствора в колонку. Если на полученной хроматограмме не обнаруживаются пики, соответствующие пикам
остаточных растворителей на хроматограммах, полученных с растворами сравнения
(а) или (б), то анализируемый образец выдерживает испытания. Если какой-либо пик
на полученной хроматограмме испытуемого раствора соответствует одному из пиков
остаточных растворителей на хроматограммах, полученных для растворов сравнения (а) или (b), и подтверждает соответствие, полученное при использовании системы А, то далее поступают следующим образом.
Подтверждение предела содержания остаточных растворителей класса 1 или класса 2
Вводят 1мл равновесной газовой фазы над раствором сравнения (с) в колонку, в
условиях, указанных для системы А или системы В, при этом высота пика,
соответствующего устанавливаемому остаточному растворителю должна быть не
менее 50% шкалы регистрирующего устройства.
Вводят 1мл равновесной газовой фазы над раствором сравнения (d) в колонку. Не должно наблюдаться никаких мешающих пиков.
1 мл равновесной газовой фазы испытуемого раствора и 1 мл газовой фазы над
раствором сравнения (с) вводят в колонку не менее трех раз.
Средняя площадь пика остаточного растворителя/растворителей на
хроматограмме равновесной газовой фазы над испытуемым раствором не должна быть больше половины площади пика остаточного растворителя/растворителей на
хроматограмме равновесной газовой фазы раствора сравнения (с). Хроматографическая система считается пригодной, если относительное
стандартное отклонение разностей площадей пиков анализируемого вещества,
полученное из трех повторных парных введений равновесной газовой фазы над раствором сравнения (а) и испытуемым раствором, не превышает 15%.
Схема проведения испытаний представлена на Рис. 2.4.24.-5.
Если уровень содержания остаточного растворителя (классов 2 или 3)
составляет 0,1% и более, для его количественного определения может быть
использован метод стандартных добавок.
# Для контроля остаточных растворителей могут быть использованы также и другие валидированные методики.
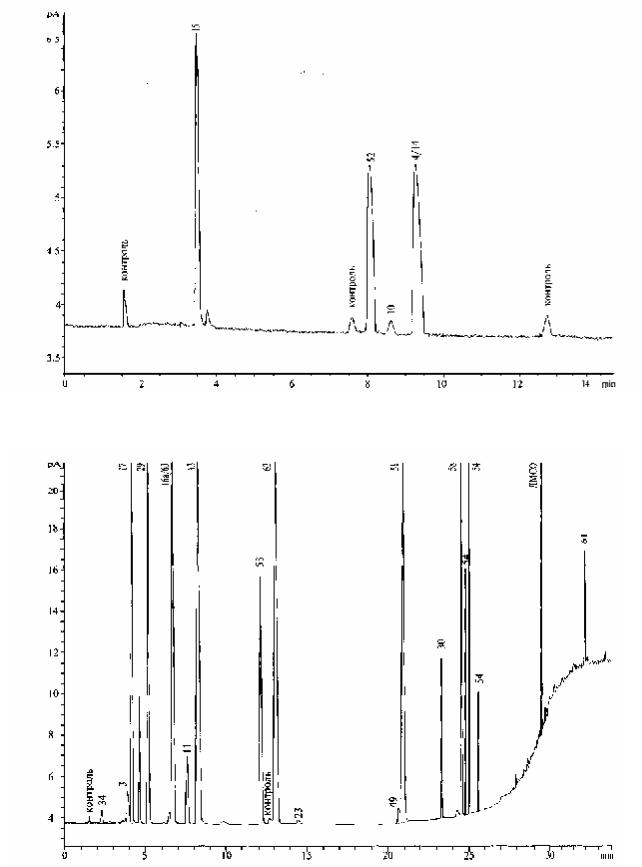
Рисунок 2.4.24.-1. Типичная хроматограмма остаточных растворителей класса 1 в условиях, описанных для системы А и пробоподготовки 1. Пламенно-ионизационный детектор.
4.бензол;10.четыреххлористый углерод;14.1,2-дихлорэтан;15.1,1-дихлорэтен;52. 1,1,1-трихлорэтан.
Рисунок 2.4.24.-2. Типичная хроматограмма остаточных растворителей класса 2 в условиях, описанных для системы А и пробоподготовки 1. Пламенно-ионизационный детектор.
3.ацетонитрил; 11.хлороформ; 13.циклогексан; 16а.цис-1,2-дихлорэтен;17.дихлорметан;29.гексан; 30.метилбутилкетон; 34.метанол; 49. пиридин; 51.толуол; 53.1,1,2-трихлорэтен; 54.орто-, мета- и пара-ксилолы; 58. хлорбензол; 61. тетралин; 62. метилциклогексан; 63. нитрометан; 64. 1,2- диметоксиэтан.