

Gosudarstvennaya_farmakopeya_RB
.pdfКАПИЛЛЯРНЫЙ ГЕЛЬ-ЭЛЕКТРОФОРЕЗ ПРИНЦИП
В капиллярном гель-электрофорезе разделение происходит внутри капилляра, заполненного гелем, который действует как молекулярное сито. Поскольку более малые молекулы легче проникают в структуру геля и мигрируют быстрее, чем большие молекулы, разделение молекул с близкими величинами
отношения заряда к массе происходит в соответствии с их размерами. Таким образом, методом капиллярного гель-электрофореза согласно величинам
молекулярных масс могут быть разделены различные биологические макромолекулы (например, белки и фрагменты ДНК), часто имеющие близкие
величины отношения заряда к массе.
ХАРАКТЕРИСТИКИ ГЕЛЕЙ
В капиллярном электрофорезе используют гели двух типов: химически модифицированные и динамически модифицированные. Химически модифицированные гели, такие как поперечно-сшитый полиакриламид, получают
полимеризацией мономеров внутри капилляра. Они обычно химически связаны с
поверхностью плавленого кварца и не могут быть удалены без разрушения капилляра. Если гели используются для анализа белков, разделяющий буфер
обычно содержит натрия додецилсульфат и пробы перед вводом денатурируют
нагреванием в смеси натрия додецилсульфата и 2-меркаптоэтанола или дитиотреитола. В случае анализа интактного антитела 2-меркаптоэтанол и дитиотреитол не применяют. Разделение с помощью поперечно-сшитых гелей может быть улучшено при изменении буферного раствора, используемого при разделении (как указано в разделе, посвящённом капиллярному зонному электрофорезу), и контролировании пористости геля во время его получения. Для
поперечно-сшитых полиакриламидных гелей пористость может быть
модифицирована путём изменения концентрации акриламида и/или пропорции сшивающего реагента. Как правило, уменьшение пористости геля приводит к
уменьшению подвижности разделяемых веществ. Вследствие жёсткости подобных
гелей может быть использован только электрокинетический ввод пробы.
Динамически модифицированные гели представляют собой гидрофильные полимеры, такие как линейный полиакриламид, производные целлюлозы, декстран
и другие, которые могут растворяться в водных буферных растворах, используемых при разделении, образуя разделяющую среду, которая также действует как
молекулярное сито. Такие разделяющие среды получить легче, чем поперечносшитые полимеры. Они могут быть приготовлены в пробирке и помещены под
давлением в капилляр с модифицированной поверхностью (без электроосмотического потока). Удаление геля после каждого ввода пробы обычно
улучшает воспроизводимость разделения. Пористость гелей может быть увеличена
при использовании полимеров с большей молекулярной массой (при определённой концентрации полимера) или путём уменьшения концентрации полимера (при определённой молекулярной массе полимера). Уменьшение пористости геля приводит к уменьшению подвижности вещества для того же самого буферного
раствора. Поскольку растворение таких полимеров в буферном растворе даёт
растворы с низкой вязкостью, может быть использован как гидродинамический, так
и электрокинетический ввод пробы.
КАПИЛЛЯРНОЕ ИЗОЭЛЕКТРИЧЕСКОЕ ФОКУСИРОВАНИЕ
ПРИНЦИП
При изоэлектрическом фокусировании молекулы мигрируют под влиянием электрического поля до тех пор, пока они остаются заряжёнными, в градиенте рН, создаваемом амфолитами, имеющими широкий диапазон значений pI
(полиаминокарбоновые кислоты), растворёнными в буферном растворе.
Тремя основными стадиями изоэлектрического фокусирования являются
загрузка, фокусирование и мобилизация.
Стадия загрузки. Могут быть использованы два способы:
-одноступенчатая загрузка: проба смешивается с амфолитами и вводится в
капилляр под давлением или с помощью вакуума;
-последовательная загрузка: в капилляр вводят вначале ведущий буферный раствор, затем амфолиты, затем пробу, смешанную с амфолитами, после чего снова амфолиты и в конце замыкающий буферный раствор. Объём пробы должен быть таким, чтобы не повлиять на величину градиента рН.
Стадия фокусирования. При наложении напряжения амфолиты, в зависимости от своего заряда, мигрируют по направлению к катоду или аноду, тем самым создавая градиент от анода (более низкие рН) к катоду (более высокие рН). Во время данной
стадии разделяемые компоненты мигрируют до тех пор, пока не достигнут рН,
соответствующего своей изоэлектрической точке, и ток не упадёт до очень низких значений.
Стадия мобилизации. Если для детекции необходима мобилизация, используют один из следующих способов:
-в первом способе мобилизация проводится в течение стадии фокусирования вследствие действия электроосмотического потока. Электроосмотический поток
должен быть достаточно малым, чтобы позволить провести фокусирование
компонентов;
-во втором способе мобилизация проводится после стадии фокусирования путём использования положительного давления;
-в третьем способе мобилизация проводится после стадии фокусирования путём добавления солей в катодный или анодный резервуар (в зависимости от направления, выбранного для мобилизации) для изменения рН в капилляре при
наложении напряжения. Поскольку рН изменяется, белки и электролиты перемещаются по направлению к резервуару, который содержит добавленные
соли, и проходят через детектор.
Достигаемое разделение, выраженное как pI, зависит от градиента рН (dpH/dx), числа амфолитов, имеющих различные величины pI, коэффициента молекулярной диффузии (D), напряжённости электрического поля (E) и того, как изменяется электрофоретическая подвижность аналита при изменении рН (- dµ/dpH):
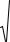
DpI = 3× |
|
D(dpH : dx) |
E(-dμ : dpH ) |
ОПТИМИЗАЦИЯ
Основными параметрами, которые следует учитывать при разработке методик разделения, являются:
Напряжение. В капиллярном изоэлектрическом фокусировании на стадии фокусирования используются электрические поля с очень высокой напряжённостью
- от 300 В/см до 1000 В/см.
Капилляр. В зависимости от способа мобилизации (см. выше)
электроосмотический поток должен быть уменьшен или подавлен. Капилляры с модифицированной поверхностью склонны уменьшать электроосмотический поток.
Растворы. Резервуар с анодным буферным раствором заполняют раствором, рН которого меньше, чем pI большинства кислотных амфолитов, а катодный резервуар
– раствором, рН которого больше, чем pI большинства основных амфолитов. Для анода часто используют фосфорную кислоту, а для катода – гидроксид натрия.
Добавление полимера, такого как метилцеллюлоза, в раствор амфолита
приводит к подавлению конвективных сил (если такие имеются) и электроосмотического потока, что обусловлено увеличением вязкости. Имеющиеся
в продаже амфолиты покрывают множество диапазонов рН, при необходимости
получить более широкий диапазон рН их можно смешивать. Широкие диапазоны рН используются для оценки величины изоэлектрической точки, в то время как более узкие применяются для улучшения точности анализа. Для градуировки можно использовать зависимость между временем миграции и изоэлектрической точкой для серии белковых маркеров.
Если необходимо, осаждение белков в изоэлектрической точке во время
стадии фокусирования может быть предотвращено с помощью добавления к
буферному раствору глицерина, поверхностно-активных веществ, мочевины или цвиттерионных буферных растворов. Следует иметь в виду, что мочевина в
зависимости от концентрации денатурирует белки.
МИЦЕЛЛЯРНАЯ ЭЛЕКТРОКИНЕТИЧЕСКАЯ ХРОМАТОГРАФИЯ (МЭКХ, MEKC) ПРИНЦИП
Вмицеллярной электрокинетической хроматографии разделение происходит
врастворе электролита, который содержит поверхностно-активное вещество в концентрации, превышающей критическую концентрацию мицеллообразования (ккм). Молекулы растворённого вещества распределяются между водным
буферным раствором и псевдонеподвижной фазой, образованной мицеллами, в соответствии со своим коэффициентом распределения. Таким образом, данный
метод можно рассматривать как гибрид электрофореза и хроматографии. Он может быть использован для разделения как заряженных, так и нейтральных веществ и
сохраняет эффективность, скорость и оборудование, присущие капиллярному электрофорезу. Одним из поверхностно-активных веществ, широко используемым в МЭКХ, является анионное поверхностно активное вещество натрия додецилсульфат. Применяются также и другие поверхностно-активные вещества, например, катионные поверхностно-активные вещества, такие как соли цетилтриметиламмония.
Механизм разделения заключается в следующем. В нейтральной или
щелочной среде возникает сильный электроосмотический поток, который перемещает ионы буферного раствора, используемого при разделении, по
направлению к катоду. Если в качестве поверхностно-активного вещества используется натрия додецилсульфат электрофоретическая миграция анионных
мицелл происходит в противоположном направлении, т.е. к аноду. В результате общая скорость миграции мицелл уменьшается по сравнению с потоком раствора электролита. В случае нейтральных разделяемых веществ, поскольку аналит способен распределяться между мицеллой и водным буферным раствором и не
имеет электрофоретической подвижности, скорость миграции аналита будет зависеть только от величины его коэффициента распределения между мицеллой и
водным буферным раствором. На электрофореграмме пики, соответствующие каждому из разделяемых незаряженных веществ, всегда находятся между пиком
маркера электроосмотического потока и пиком мицеллы (время между этими двумя пиками называется окном разделения). Для заряженных разделяемых веществ
скорость миграции зависит как от коэффициента распределения вещества между мицеллой и водным буферным раствором, так и от электрофоретической подвижности вещества в отсутствии мицеллы.
Поскольку механизм разделения в МЭКХ для нейтральных и слабо ионизированных веществ, главным образом, хроматографический, миграция
вещества и разрешение могут быть охарактеризованы с помощью фактора удерживания вещества (k), также известного как концентрационный коэффициент
распределения (Dm ), который представляет собой отношение количество
разделяемого вещества, находящегося в мицелле, к количеству данного вещества в подвижной фазе.
Для нейтральных соединений k определяется как:
k = |
|
tR - t0 |
|
= K ´ |
VS |
, |
||
|
æ |
|
tR |
ö |
|
|||
|
t0 |
|
VM |
|||||
|
´ ç1 |
- |
|
÷ |
|
|
|
|
|
|
|
|
|||||
|
|
è |
|
tmc ø |
|
|
|
|
где:
tR – время миграции вещества,
t0 – время анализа неудерживаемого соединения (определяемое с помощью маркера электроосмотического тока, не входящего в мицеллу, например, метанола),
tmc – время миграции мицеллы (изменяется с помощью маркера мицеллы, такого как Судан III, который мигрирует, полностью находясь в мицеллах),
K – коэффициент распределения вещества, VS – объём мицеллярной фазы,
VM – объём подвижной фазы.
Аналогично, разрешение пиков двух близко мигрирующих веществ
определяется как:
|
|
|
|
α -1 |
|
|
|
|
|
1- çæ |
t0 |
÷ö |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Rs = |
|
N |
× |
× |
kb |
|
× |
|
è tm ø |
|
, |
||||
4 |
α |
kb +1 |
|
|
æ t0 |
ö |
|||||||||
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
1 |
+ ka ×ç |
|
÷ |
|
|||
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
è tmc ø |
|
|||

где:
N – число теоретических тарелок для одного из веществ, α – селективность,
ka и kb – факторы удерживания для обоих веществ соответственно (kb> ka).
Подобные, но не идентичные, уравнения для значений k и Rs используются и
в случае разделения заряженных веществ.
ОПТИМИЗАЦИЯ
Основными параметрами, которые следует принимать во внимание при
разработке методик разделения в МЭКХ, являются инструментальные параметры, а также параметры раствора электролита.
Инструментальные параметры
Напряжение. Время разделения обратно пропорционально приложенному
напряжению. Однако увеличение напряжения может вызвать избыточное выделение тепла, повышение температуры и, как результат, образование градиентов температуры и вязкости в буферном растворе, находящемся в капилляре. Данный эффект особенно характерен для буферных растворов с высокой электропроводностью и содержащих мицеллы. Неудовлетворительная тепловая конвекция приводит к расширению полосы и уменьшает разрешение.
Температура. Изменения температуры капилляра влияют на коэффициент
распределения вещества между буферным раствором и мицеллой, на критическую концентрацию мицеллообразования и вязкость буферного раствора. Данные
параметры вносят вклад в величину времени удерживания вещества.
Использование хорошей охлаждающей системы улучшает воспроизводимость времен миграции веществ.
Капилляр. Как и в случае капиллярного электрофореза со свободным раствором размеры капилляра (длина и внутренний диаметр) влияют на время анализа и эффективность разделения. Увеличение как эффективной, так и общей длины капилляра может ослабить электрическое поле (в случае работы при
постоянном напряжении), увеличить время миграции и улучшить эффективность
разделения. Внутренний диаметр контролирует тепловую конвекцию (для определённого буферного раствора и электрического поля) и, соответственно,
расширению полосы вещества.
Параметры раствора электролита
Тип и концентрация поверхностно-активного вещества. Тип поверхностно-
активного вещества таким же образом, как и неподвижная фаза в хроматографии, влияет на разрешение, поскольку изменяет селективность разделения. Величина lg k нейтрального соединения линейно увеличивается при увеличении концентрации поверхностно-активного вещества в подвижной фазе. Поскольку разрешение в
МЭКХ достигает максимума при приближении величины k к 
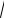 tmc : t0 , изменение
tmc : t0 , изменение
концентрации поверхностно-активного вещества в подвижной фазе изменяет величину разрешения.
Величина рН буферного раствора. Несмотря на то что величина pH не изменяет коэффициент распределения неионизированных веществ, она может
изменять электроосмотический поток в капиллярах с немодифицированной
поверхностью. Уменьшение рН буферного раствора уменьшает электроосмотический поток и, тем самым, увеличивает разрешение нейтральных
веществ в МЭКХ вследствие увеличения времени анализа.
Органические растворители. Для улучшения МЭКХ-разделения
гидрофобных соединений к раствору электролита могут быть добавлены органические модификаторы (метанол, пропанол, ацетонитрил и другие). Добавление этих модификаторов обычно уменьшает время миграции и селективность разделения. Поскольку добавление органических модификаторов
влияет на величину критической концентрации мицеллообразования, определённая концентрация поверхностно-активного вещества может быть использована только в
пределах некоторого процентного содержания органического модификатора до прекращения мицеллообразования или отрицательного влияния на этот процесс,
приводящего к исчезновению мицелл и прекращению разделения. Диссоциация мицелл в присутствии большого количества органического растворителя не всегда
означает невозможность дальнейшего разделения; в некоторых случаях гидрофобное взаимодействие между мономером ионного поверхностно-активного вещества и нейтральным разделяемым веществом приводит к образованию сольвофобных комплексов, которые могут быть разделены электрофоретически.
Добавки для хиральных разделений. Для разделения энантиомеров с
использованием МЭКХ хиральный селектор включается в мицеллярную систему либо ковалентно связывается с поверхностно-активным веществом, либо
добавляется к раствору электролита, в которм происходит разделение мицелл.
Мицеллы, способные к хиральным разделениям, обычно включают в себя соли N- додеканоил-L-аминокислот, соли желчных кислот и т.д. Хиральное разделение может быть также проведено с помощью циклодекстринов, добавленных к растворам электролитов, содержащих мицеллы ахиральных поверхностноактивных веществ.
Другие добавки. Существует ряд подходов, предполагающих добавление
различных реагентов к буферному раствору для изменения селективности.
Добавление некоторых типов циклодекстринов к буферному раствору также может быть использовано для уменьшения взаимодействия гидрофобных веществ с
мицеллой, тем самым увеличивая селективность разделения для такого типа
соединений.
Добавление веществ, которые способны изменять взаимодействия между разделяемым веществом и мицеллой вследствие адсорбции на последней,
используется для улучшения селективности разделения в МЭКХ. Данные добавки могут представлять собой второе поверхностно-активное вещество (ионное или
неионное), которое увеличивает количество смешанных мицелл или катионов металлов, которые растворяются в мицелле и образуют координационные
комплексы с разделяемыми веществами.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Пики должны быть разделены соответствующими временами миграции,
чтобы в итоге получить исправленную площадь, позволяющую:
- компенсировать смещения времен удерживания от анализа к анализу, тем самым уменьшить различия в величине отклика,
-компенсировать различные отклики компонентов пробы с различными временами
миграции.
При использовании внутреннего стандарта следует убедиться в том, что пик
исследуемого вещества не скрыт аналогичным пиком внутренного стандарта.
РАСЧЁТЫ
По полученным данным рассчитывают содержание исследуемого компонента или компонентов. Если требуется, рассчитывают процентное содержание одного
или нескольких компонентов образца путём определения исправленной площади пика или пиков, как процентной части исправленных площадей всех пиков,
исключая пики, соответствующие растворителям или любым добавленным реагентам (метод нормализации). Рекомендуется использование автоматической
системы интеграции (интегратора или системы получения и обработки данных)
ПРИГОДНОСТЬ СИСТЕМЫ
Параметры пригодности системы используются для проверки поведения системы капиллярного электрофореза. Выбор этих параметров зависит от используемой разновидности капиллярного электрофореза. К ним относятся:
фактор удерживания (k) (только для мицеллярной электрокинетической
хроматографии), число теоретических тарелок (N), фактор симметрии (As) и разрешение (Rs). В предыдущих разделах были описаны уравнения для расчётов N
и Rs, ниже приведены уравнения, позволяющие рассчитать эти параметры из
электрофореграмм.
ЧИСЛО ТЕОРЕТИЧЕСКИХ ТАРЕЛОК
Число теоретических тарелок (N) может быть рассчитано с помощью выражения:
æ tR |
ö |
2 |
|
N = 5,54ç |
|
÷ |
, |
|
|||
è wh ø |
|
||
где: |
|
|
|
tR – время миграции или расстояние по базовой линии от точки ввода пробы до
перпендикуляра, опущенного из максимума пика, соответствующего компоненту, wh – ширина пика на половине высоты.
РАЗРЕШЕНИЕ
Разрешение (Rs) близких по высоте пиков двух компонентов может быть рассчитано из выражения:
Rs = |
1,18(tR |
− tR ) |
|
|
2 |
2 |
|
|
|
||
|
wh |
+ wh |
|
|
1 |
2 |
|
tR2 > tR1 ,

где:
tR1 и tR2 – времена миграции или расстояния по базовой линии от точки ввода пробы до перпендикуляров, опущенных из максимумов двух соседних пиков,
wh1 and wh2 – ширина пиков на половине высоты.
При необходимости может быть измерена высота седловины (Hv) между
двумя частично разделёнными пиками в стандартном образце, а также высота меньшего пика (Hp) и рассчитана величина коэффициента разделения неполностью разделённых пиков:
p= H p v H v
ФАКТОР СИММЕТРИИ
Фактор симметрии пика (As) рассчитывается из выражения:
As = w0,05
2d
w0,05 – ширина пика на одной двадцатой высоты,
d – расстояние между перпендикуляром, опущенным из максимума пика и передним
краем пика на одной двадцатой высоты.
Испытания на повторяемость площадей (стандартное отклонение площадей
или отношений площадь/время миграции) и времен миграции (стандартное
отклонение времени миграции) используются в качестве параметров пригодности. Повторяемость времени миграции обеспечивает испытание методики промывки
капилляра. Альтернативным способом предупреждения ухудшения повторяемости
времени миграции является использование отношения времени миграции относительно внутреннего стандарта.
Для определения родственных соединений может быть полезно испытание на подтверждение отношения сигнал/шум для стандартного образца (или определение предела количественного определения).
ОТНОШЕНИЕ СИГНАЛ/ШУМ
Пределы обнаружения и количественного определения отвечают отношению сигнал/шум, соответственно, 3 и 10. Отношение сигнал/шум (S/N) рассчитывается
из выражения:
NS = 2hH ,
где:
H – высота пика, соответствующая рассматриваемому компоненту, на
электрофореграмме, полученной при использовании рекомендованного раствора
сравнения. Высота пика измерена от максимума пика до экстраполированной базовой линии для сигнала, величина которого эквивалентна двадцатикратному
превышению ширины пика на половине высоты,
h – размах для фонового шума (уровень шума) на электрофореграмме, полученной
при введении контрольной пробы. Величина эквивалентна двадцатикратному превышению ширины пика на половине высоты для пика на электрофореграмме,
полученной при использовании рекомендованного раствора сравнения и, если возможно, расположенного на одном и том же расстоянии от места возможного
обнаружения пика.
2.2.48. РАМАНОВСКАЯ СПЕКТРОМЕТРИЯ (# СПЕКТРОМЕТРИЯ КОМБИНАЦИОННОГО РАССЕЯНИЯ)
Рамановская спектрометрия (неупругое рассеяние света) – это процесс рассеяния света, при котором исследуемый образец облучается интенсивным
монохроматическим светом (обычно излучение лазера) и для света, рассеиваемого образцом, определяется сдвиг частоты.
Рамановская спектрометрия является дополнительным методом по отношению к ИК-спектрометрии, в том смысле, что оба этих метода основаны на
молекулярных колебаниях частиц исследуемого вещества. Однако рамановская и ИК-спектрометрия отличаются по относительной чувствительности для различных функциональных групп. Рамановская спектрометрия особенно чувствительна к
неполярным связям (например, одинарные или кратные связи C-C), тогда как вода,
для которой характерно сильное поглощение в ИК-области, обладает слабым
рамановским рассеянием и поэтому хорошо подходит на роль растворителя в рамановской спектрометрии.
Прибор. Спектрометры для получения рамановских спектров обычно состоят
из следующих компонентов:
-источник монохроматического света, чаще всего лазер с длиной волны в ультрафиолетовой, видимой или ближней инфракрасной области,
-подходящая оптика (линзы, зеркала, световоды), которые направляют возбуждающее излучение на образец и собирают свет, рассеиваемый образцом,
-оптическое устройство (монохроматор или фильтр), которое пропускает
сдвинутое по частоте рамановское рассеяние и препятствует попаданию на
детектор интенсивного света, частота которого равна частоте возбуждающего света (рэлеевское рассеяние),
-диспергирующее устройство (дифракционная решётка или призма), соединённое
со щелью, регулирующей длину волны, и детектором (обычно фотоумножитель),
или:
- диспергирующее устройство (дифракционная решётка или призма), соединённое
с многоканальным детектором (обычно прибор с зарядовой связью (ПЗС, CCD)), или:
- интерферометр, регистрирующий интенсивность рассеянного света во времени, и устройство обработки данных, которое с помощью фурье-преобразования
преобразует данные в диапазон частот или волновых чисел.
ПОДГОТОВКА ОБРАЗЦА
Рамановские спектры могут быть получены для твёрдых веществ, жидкостей
и газов, причём как напрямую, так и помещенных в стеклянные контейнеры или трубки, как правило, без предварительной подготовки образца или растворения.
Основным ограничением рамановской спектрометрии является флуоресценция примесей, которая мешает детектору улавливать значительно
более слабый рамановский сигнал. Флуоресценции можно избежать при использовании в качестве возбуждающего света лазерного излучения с большой длиной волны, например, находящейся в ближней инфракрасной области. Интенсивность некоторых рамановских линий может быть усилена несколькими
способами, например, путём использования резонансной рамановской спектрометрии (RR) или усиленной поверхностью рамановской спектрометрии
(SERS).
Из-за узости светового потока падающего лазерного излучения, для
получения спектра необходимо всего лишь несколько микролитров образца. Тем не менее, следует учитывать неоднородность образца, если только его объём не
увеличен, например, путём вращения образца.
ИДЕНТИФИКАЦИЯ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ С ИСПОЛЬЗОВАНИЕМ
ВЕЩЕСТВ СРАВНЕНИЯ
Исследуемое вещество и вещество сравнения подвергают одинаковой
обработке и затем при одинаковых условиях получают их спектры. Максимумы в спектре исследуемого вещества совпадают по положению и интенсивности с
соответствующими максимумами в спектре вещества сравнения (CRS).
Если спектры, полученные для твёрдых веществ, имеют различия в положениях максимумов, исследуемое вещество и вещество сравнения подвергают одинаковой обработке для того, чтобы они выкристаллизовались или образовались в одинаковой форме, либо поступают так, как описано в частной статье, и затем снимают спектры.
Поскольку закон Бера-Ламберта для рамановской спектроскопии не
выполняется, интенсивность рамановского излучения прямо пропорциональна
концентрации рассеивающих частиц. Как и в случае других спектроскопических методов количественное определение может быть проведено с использованием
известных количеств или концентраций веществ сравнения. Вследствие
небольшого пространственного разрешения метода следует обращать внимание на качество исследуемых образцов и образцов сравнения, например, необходимо быть уверенным в том, что они находятся в одном и том же физическом состоянии,
либо использовать внутренний стандарт для жидких образцов.
ИДЕНТИФИКАЦИЯ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ С ИСПОЛЬЗОВАНИЕМ СПЕКТРАЛЬНЫХ БИБЛИОТЕК И СТАТИСТИЧЕСКИХ МЕТОДОВ
КЛАССИФИКАЦИИ И ГРАДУИРОВКИ
Контроль за работой прибора. При использовании прибора следуют
инструкциям производителя; регулярно, в зависимости от применения прибора и
исследуемых веществ, проводят предписанные градуировки и испытания работы системы. При использовании рамановской спектрометрии для количественных определений либо при создании спектральных библиотек сравнения для (хемометрической) классификации или градуировки необходимо обращать особое
внимание на то, чтобы были сделаны все поправки в измерения либо предприняты