

Bioregenerative Engineering Principles and Applications - Shu Q. Liu
..pdf676 |
VASCULAR REGENERATIVE ENGINEERING |
|
|
|
Fibroatheroma, Late-Stage Necrosis |
|
|
|
|
A |
B |
C |
D |
E |
|
NC |
|
|
|
|
Macrophages |
Glycophorin A |
Iron |
vWF |
Thin-Cap Fibroatheroma |
|
|
|
|
F |
G |
H |
I |
J |
Macrophages |
Glycophorin A |
Iron |
vWF |
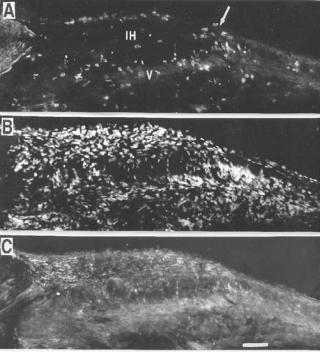
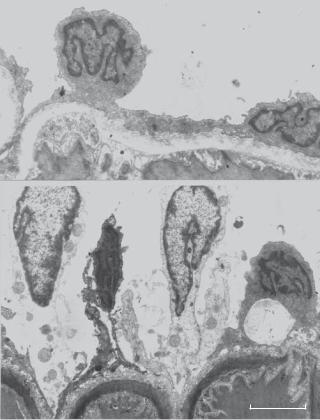
Figure 15.3. Pathological changes in human late-stage necrotic fibroatheroma (panels A–E) and thin-cap fibroatheroma (panels F–J). Panel A shows a low-power view of a fibroatheroma with a late-stage necrotic core (NC) (Movat pentachrome, ×20). Panel B shows intense staining of CD68positive macrophages within the necrotic core (×200). Panel C shows extensive staining for glycophorin A in erythrocyte membranes localized with numerous cholesterol clefts within the necrotic core (×200). Panel D shows iron deposits (blue pigment) within foam cells (Mallory’s stain ×200). Panel E shows microvessels bordering the necrotic core with perivascular deposition of von Willebrand factor (vWF) (×400). Panel F shows a low-power view of a fibroatheroma with a thin fibrous cap (arrow) overlying a relatively large necrotic core (Movat pentachrome, ×20). The fibrous cap is devoid of smooth muscle cells (not shown) and is heavily infiltrated by CD68-positive macrophages (panel G, ×200). Panel H shows intense staining for glycophorin A in erythrocyte membranes within the necrotic core, together with cholesterol clefts (×100). Panel I shows an adjacent coronary segment with iron deposits (blue pigment) in a macrophage-rich region deep within the plaque (Mallory’s stain, ×200). Panel J shows diffuse, perivascular deposits of von Willebrand factor in microvessels, indicating that leaky vessels border the necrotic core (×400). (Reprinted with permission from Kolodgie FD et al: Intraplaque hemorrhage and progression of coronary atheroma, New Engl J Med 349:2316–25, copyright 2003 Massachusetts Medical Society. All rights reserved.)
takes a considerably long time, ranging from years to decades, atherosclerosis is often detected in aged population (> 40 years old). Grown atheroma can partially or completely block the lumen of arteries, resulting in ischemia or infarction of distal tissues or organs. Atherosclerosis often involves the brain (causing stroke), the heart (cardiac ischemia and infarction), kidney (renal infarction), and lower limbs (ischemia). Atherosclerosis is the leading cause of human death.
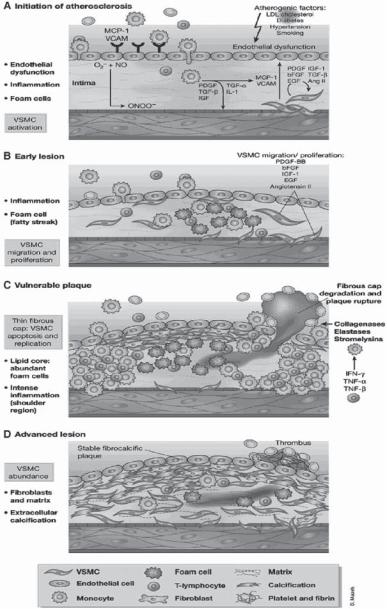
The pathogenesis of atherosclerosis is related to various types of endothelial injury. A number of factors have been found to induce endothelial injury, including bacterial and viral infection, physical trauma (surgery and catheterization), chemical toxins, bloodflowrelated shear and tensile forces, and physical trauma. These factors induce endothelial cell injury or detachment and inflammatory reactions in the wall of blood vessels (Fig. 15.4). Endothelial cell injury may result in changes in endothelial permeability and adhesiveness to blood cells, and the release of growth factors and cytokines. Altered endothelial permeability may facilitate the transport of cholesterol and low-density lipoproteins across the endothelium. Altered endothelial adhesiveness may promote leukocyte attachment to and migrate through the endothelium. Released growth factors and cytokines may attract more
Blood velocity profile
A
B
Blood vessel
Cylindrical implant |
|
C |
|
0° |
|
109° |
|
180° |
|
0° |
D |
E |
0° |
55° |
|
55° |
||
|
180°
180°
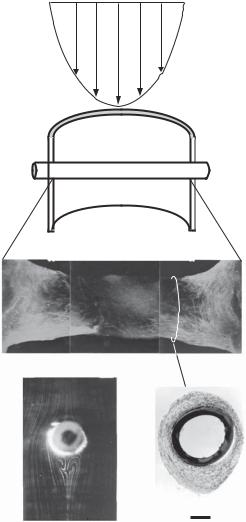
Figure 15.6. Influence of blood flow and shear stress on thrombosis, intimal hyperplasia, and the migration of vascular smooth muscle cells on a cylindrical polymer tube implanted into the rat aorta.
(A) Parabolic profile of bloodflow. (B) Schematic representation of the polymer implant in the aorta.
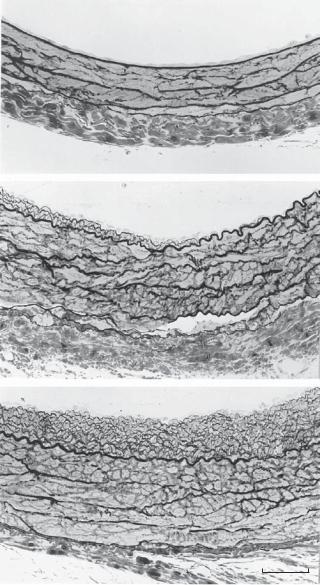
(C) Thrombotic tissue on the implanted polymer tube with smooth muscle cells (green in color, labeled with an anti-smooth muscle α-actin antibody), which migrated from the wall of the host aorta. Note that the maximal bloodflow velocity is associated with a minimal level of thrombosis in the central region of the implanted polymer tube. In this model, the implanted cylindrical tube is subject to a laminar bloodflow with the tube axis perpendicular to the bloodflow. The interaction of the implanted cylindrical tube with the bloodflow results in a unique distribution of fluid shear stress on the surface of the implanted tube in its circumferential direction: a leading stagnation point at 0º (with zero shear stress), an increasing shear stress region from 0º to 55º with a maximal shear stress point at 55º, a decreasing shear stress region from 55º to 109º, a flow separation point at 109º (with zero shear stress), a vortex flow region from 109º to 180º, and a trailing stagnation point at 180º (with zero shear stress). The flow pattern is symmetric from 0º to −180º. The distribution of fluid shear stress on the surface of the implanted cylindrical tube can be estimated by using an fluid dynamic model. The level of fluid shear stress is inversely correlated to the migration of the vascular smooth muscle cells. As shown in panel C, the maximal shear stress point at 55º is associated with minimal migration of smooth muscle cells. (D) Flow pattern around the implanted cylindrical polymer tube detected in an flow simulation model with a physiological Reynolds number in vitro. Note the formation of vortex flow behind the polymer tube. (E) A histological micrograph derived from a transverse section prepared from the implanted polymer tube with encapsulating thrombotic tissue as indicated in panel C. Note that the minimal shear stress at the leading and trailing stagnation points (0º and 180º) is associated with maximal thrombosis and intimal hyperplasia whereas the maximal shear stress at 55º is associated with minimal thrombosis and intimal hyperplasia. Scale: 100 μm.
682 VASCULAR REGENERATIVE ENGINEERING
Hyperlipidemia is induced by two mechanisms: excessive intake of lipids (fatty acids and cholesterol) and reduced clearance of plasma low-density lipoproteins, which carry lipids. The later is considered a disorder. Hyperlipidemia is divided into two groups, based on the pathogenesis of the disorder: primary and secondary. The primary form is a hereditary disorder and is related to genetic defects. In some patients, this is due to a single gene defect, which is called monogenic hyperlipidemia. This type of disorder can be predicted on the basis of the Mendelian genetic trait. In others, it is caused by a combination of multiple gene mutations and environmental insults, which is called polygenic hyperlipidemia. In this case, the plasma cholesterol level of the majority members of a family may go up some time during the lifespan, if the intake of saturated fats and cholesterol is high. Usually, the polygenic type prevails in the human population. Secondary hyperlipidemia is a complication of metabolic disorders, such as diabetes. A common sign of hyperlipidemia is the elevation of plasma cholesterol and triglyceride.
Atherosclerosis is characterized by several stages of progressive pathological lesions, including the fatty streak, the intermediate, and the fibrous plaque stages. During the fatty streak stage, monocytes and T lymphocytes migrate into and accumulate in the intima. The monocytes are transformed into macrophages, which convert to foam cells. The fatty streaks are often found at locations such as curved arteries and bifurcations. During the intermediate stage, atherosclerotic lesions often contain several types of cell, including foam cells, T lymphocytes, and smooth muscle cells, and loosely organized extracellular matrix components, including collagen and elastin fibers. During the fibrous plaque stage, the density of smooth muscle cells increases together with excessive production of extracellular matrix. Foam cells and T lymphocytes as well as lipid components are found in atheroma. These components are usually capped with a layer of connective tissue. Since the lesion increases in size during this stage, clinical consequences such as vascular stenosis and occlusion often occur. Furthermore, atherosclerotic lesions may detach from the wall of blood vessels, resulting in arterial embolism.
Atherosclerosis is associated with cell apoptosis in humans and animal models. Apoptotic cells are often found at the base of atheroma. Several death regulatory factors, including Bcl-2, caspase 1 and 3, and Fas receptor are upregulated in atheroma, suggesting a role of these death factors in the regulation of cell apoptosis. The activation of apoptosis in atherosclerotic lesions may be a self-defense mechanism that is initiated in response to increased smooth muscle cell proliferation. However, this process may reduce the stability of atherosclerotic plaques, leading to formation of emboli.
Animal Models of Atherosclerosis [15.8]. Atherosclerosis models can be established in the mouse, rat, guinea pig, rabbit, dog, pig, and nonhuman primate. Several approaches can be used to create an atherosclerosis model, including high-cholesterol diet feeding, genetic modulation, and vascular injury. High-cholesterol-diet feeding induces hypercholesterolemia and fatty streak-like lesions in the arterial system. Although fatty streaks do not represent human atherosclerosis, they are similar in structure to early atherosclerotic lesions in humans. Rabbits are often used for this model since these animals are susceptible to hypercholesterolemia and develop fatty streak-like lesions within a short period.
Genetic modulation of proteins that regulate lipid metabolism can render animals vulnerable to atherosclerosis. A typical model is experimental atherosclerosis due to apolipoprotein E (apoE)-deficiency. ApoE is a protein that plays a role in the clearance of blood lipid molecules, which contribute to atherogenesis. The deficiency of apoE enhances the
684 VASCULAR REGENERATIVE ENGINEERING
hypertension, diabetes, and obesity. These disorders facilitate the initiation and development of atherosclerosis. A number of methods have been established and used for the prevention of atherosclerosis. These include diet control, appropriate daily exercise, and administration of antioxidant agents. For diet control, Low-fat, low-cholesterol, and soybean-rich foods are beneficial. Daily exercise is also effective for the prevention of atherogenesis. While the mechanisms are poorly understood, exercise-enhanced bloodflow and increased fluid shear stress may reduce the rate of atherogenesis. Experimental investigations have demonstrated that an increase in blood shear stress with respect to the physiological level exerts an inhibitory effect on the activity of growth factors and the proliferation and migration of vascular smooth muscle cells. These mechanisms may contribute to shear stress-related suppression of atherogenesis. Furthermore, the administration of antioxidants, such as vitamin E, vitamin C, vitamin A, and β-carotene (a precursor of vitamin A), may help to prevent atherogenesis. The oxidation of low-density lipoproteins (LDL) facilitates macrophage endocytosis of LDL, enhances macrophage conversion to foam cells, and contributes to atherogenesis. A treatment with antioxidant agents can reduce the level of LDL oxidation, thus reducing atherogenic activities.
Conventional Treatment of Atherosclerosis [15.9]. When atherosclerosis is diagnosed, it can be treated with several approaches, including the administration of antihyperlipidemia agents, anti-SMC proliferation agents, and vasodilators, reduction in cardiac workload, angioplasty, vascular stenting, and vascular reconstruction. These approaches are described as follows.
Antihyperlipidemia Agents. Common anti-hyperlipidemia agents include 3-hydroxy-3- methylglutaryl coenzyme A (HMG CoA) reductase inhibitor, fibric acids, and bile acidbinding resins. 3-Hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitor is a fungus-derived compound that inhibits HMG CoA reductase, a rate-controlling enzyme for the synthesis of cholesterol. Fibric acids are substances that reduce the plasma level of very-low-density lipoproteins, low density lipoproteins, and intermediate density lipoproteins. Bile acid-binding resins are anion exchange polymeric resins that facilitate the removal of cholesterol from the intestinal system. All these agents can be used to lower the level of lipids.
Antiproliferative Agents. Several types of agents have been developed and used for suppressing smooth muscle cell proliferation, including inhibitors for angiotensin converting enzyme (ACE), angiotensin II type 1 receptor, and growth factors. Angiotensin converting enzyme catalyzes the cleavage of angiotensin I and converts angiotensin I to angiotensin II, which not only stimulates smooth muscle contraction but also promotes smooth muscle proliferation. The suppression of ACE reduces smooth muscle proliferation. Common ACE inhibitors include captopril, enalapril, and lisinopril.
The effect of angiotensin II on the proliferative activity of vascular smooth muscle cells depends on its interaction with the angiotensin II type 1 receptor on the cell membrane. The blockage of this type of receptor can significantly suppress angiotensin II-induced smooth muscle proliferation. Most angiotensin II antagonists are peptide-based angiotensin II analogues, which have a structure similar to that of angiotensin II with a modified amino acid sequence. These peptides compete with angiotensin II for receptor binding, but do not induce smooth muscle activation and, therefore, can reduce the mitogenic effect of angiotensin II.
VASCULAR DISORDERS |
685 |
Growth factors, such as platelet-derived growth factor, stimulate smooth muscle proliferation and migration. The suppression of growth factors reduces smooth muscle mitogenic activities. A typical antagonist for platelet-derived growth factor is terbinafine. This agent has been shown to inhibit the activity of platelet-derived growth factor and thus suppress cell proliferation in experimental models of atherosclerosis. Other antiproliferative agents include heparin, rapamycin, and Taxol. Heparin is an anticoagulant and also exerts an inhibitory effect on smooth muscle proliferation. Rpamycin is a natural antibiotic found in a microorganism Streptomyxes hygroscopius. This agent inhibits the progression of cell cycle and thus suppresses smooth muscle proliferation. Taxol is a compound extracted from the bark of Taxus brevifolia, a Pacific yew. Taxol has been found to stabilize microtubules, suppress cell division, and induce cell apoptosis. When applied to blood vessels, Taxol inhibits SMC proliferation and prevent intimal hyperplasia.
Treatment of Angina Due to Ischemia. When angina occurs due to ischemia, vasodilators can be used to increase bloodflow to the heart, thus reducing ischemia. Two types of drug are commonly used for such a purpose: organic nitrates and calcium channel blockers. Organic nitrates induce relaxation of coronary arterial and venous smooth muscle cells, resulting dilation of these blood vessels and an increase in bloodflow. Commonly used nitrates include amyl nitrite, nitroglycerin, and isosorbide dinitrate. Calcium channel blockers can be used to reduce the release of calcium. Calcium is necessary for the initiation of vascular smooth muscle contraction and is released into the cytoplasm through its channels. The blockade of calcium channels leads to a reduction in the contractility of smooth muscle cells. Commonly used agents in this class include nicardipine, nifedipine, nimodipine, and verapamil.
Reduction in Cardiac Workload. A decrease in cardiac workload is beneficial to the treatment of ischemia and prevention of cardiac infarction. β-adrenergic antagonists are effective agents for such a purpose. Adrenergic agents, including epinephrine and norepinephrine, enhance myocardial contractility and, therefore, induce an increase in the workload and oxygen consumption of the heart. β-adrenergic antagonists, such as atenolol, acebutolol, metoprolol, nadolol, pindolol, propranolol, and timolol, can block the β- adrenergic receptors, thus reducing cardiac contractility and workload.
Treatment of Malfunctioned Arteries with Angioplasty. For advanced atherosclerosis with clinical consequences, such as vascular stenosis and occlusion, surgical interventions are necessary. One surgical approach is angioplasty, by which stenotic arteries are reopened by inserting a balloon-tipped catheter into and expand the stenotic arteries by balloon inflation. This approach is relatively simple compared to other types of surgical interventions, such as stenting and grafting. However, mechanical expansion induces stretch and injury of the arterial wall, which initiates blood coagulation, thrombogenesis, and intimal hyperplasia. Restenosis occurs after angioplasty in a large fraction of patients. Thus, angioplasty is primarily used for temporary relief of arterial occlusive disorders.
Treatment of Malfunctioned Arteries with Stents. Arterial stenting is a procedure by which a cylindrical wire-frame is inserted into a stenotic artery with the aid of a balloontipped catheter. The wire-frame is expanded by inflating the balloon. The balloon and catheter are then retreated, leaving the opened wire-frame within the lumen so that the artery is kept open. Although this is a relatively simple procedure, stenting often induces