

Neutron Scattering in Biology - Fitter Gutberlet and Katsaras
.pdf10 Structure and Kinetics of Proteins |
203 |
10.5 Conclusions and Outlook
Small-angle scattering is a convenient and powerful tool for the investigation of protein–protein and protein–nucleic acid interactions. The lower flux of neutron sources compared to the high brilliance of third-generation X-ray sources is partially compensated by the higher contrast (= scattering power) of proteins in D2O. The main advantages of (“cold”) neutrons compared to X-rays are the absence of radiation damage and the benefits of contrast variation. Solvent contrast-variation and specific deuteration allow one to dissect biological macromolecules into partial structures. Novel approaches for the analysis of small-angle scattering data using these techniques are promising a rapid structure determination in the low-resolution regime [42]. Time-resolved neutron small-angle scattering is emerging as a tool for unique information concerning the structure of products and intermediates in the reaction of proteins. Since the derived kinetic data can be directly pinned down to specific conformational rearrangement processes, time-resolved small angle scattering permits the structural analysis of single reaction steps in protein interactions. Complete sets of such kinetic data would be an important step towards systems biology. The recent development of high-count-rate neutron detectors and of neutron-focusing optics will permit to measure the structures and kinetics of smaller molecules. The advent and further improvement of new pulsed neutron sources with higher fluxes will in future allow one to study more rapid reactions of macromolecules in solution and/or to use less of the sometimes very precious material.
References
1.H. Frauenfelder, S.G. Sligar, P.G. Wolynes, Science 254, 1598–1603 (1991)
2.A.E. Garcia, J.A. Krumhansl, H. Frauenfelder, Proteins 29, 153–160 (1997)
3.D.I. Svergun, M.H.J. Koch, Rep. Progr. Phys. 66, 1735 (2003)
4.D.I. Svergun, M.H.J. Koch, Curr. Opin. Struct. Biol. 12, 654 (2002)
5.O. Glatter, J. Appl. Cryst. 10, 415–421 (1997)
6.H. Lederer, R.P. May, J.K. Kjems, W. Schaefer, H.L. Crespi, H. Heumann, Eur.
J.Biochem. 156, 655–659 (1986)
7.R. Stegmann, E. Manakova, M. Roessle, H. Heumann, S.E. Nieba-Axmann, A. Pluckthun, T. Hermann, R.P. May, A. Wiedenmann, J. Struct. Biol. 121, 30–40 (1998)
8.K. Mo at, Acta Cryst. A 54, (1998) 833–841
9.I. Schlichting, Accounts Chem. Res. 33, 532–538 (2000)
10.B. Perman, V. Srajer, Z. Ren, T. Teng, C. Pradervand, T. Ursby, D. Bourgeois,
F.Schotte, M. Wul , R. Kort, K. Hellingwerf, K. Mo at, Science 279, 1946– 1950 (1997)
11.B. Perman, S. Anderson, M. Schmidt, K. Mo at, Cell. Mol. Biol. 46, 895–913 (2000)
12.M.H. Chestnut, D.P. Siegel, J.L. Burns, Y. Talmon, Microscopy Res. Tech. 20, 95–101, (1992)
204 M.W. Roessle et al.
13.E.M. Mandelkow, E. Mandelkow, R.A. Milligan, J. Cell Biol. 114, 977–991 (1991)
14.H.R. Saibil, Nat. Struct. Biol. 7, 711–714 (2000)
15.A.K. Bhuyan, J.B. Udgaonkar, Proteins 32, 241–247 (1998)
16.S. Cusack, Neutron and Synchroton Radiation for Condensed Matter Studies, Les editions de physique, vol. III (Springer Verlag, 1994)
17.S.A. Endow, Nat. Cell Biol. 1, 163–167 (1999)
18.R.D. Vale, R.A. Milligan, Science 288, 95–100 (2000)
19.K.Jr. Kinosita, FASEB J. 13, 201–208 (1999)
20.A. Mattevi, M. Rizzi, M. Bolognesi, Curr. Opin. Struct. Biol. 6, 824–829 (1996)
21.K.E. Neet, Methods Enzymol. 249, 519–567 (1995)
22.J. Monod, J. Wyman, J.P Changeux, J. Mol. Biol. 12, 88–118 (1965)
23.M.F. Perutz, Quart. Rev. Biophys. 22, 139–237 (1989)
24.R.J. Ellis, Nature 328, 378–379 (1987)
25.T. Langer, G. Pfeifer, J. Martin, W. Baumeister, F.U. Hartl, EMBO 11, 4757 (1992)
26.M. Klumpp, W. Baumeister, FEBS Lett. 430, 73–77 (1998)
27.M. Roessle, E. Manakova, I. Lauer, T. Nawroth, J. Holzinger, T. Narayanan,
S.Bernstor , H. Amenitsch, H. Heumann, J. Appl. Cryst. 33, 548–551 (2000)
28.A. Neidhart, T. Nawroth, M. Huetsch, K Dose, FEBS Lett. 280, 179–182 (1991)
29.M. Roessle, E. Manakova, I. Lauer, T. Nawroth, R. Gebhardt, T. Narayanan,
H.Heumann, ESRF Newslett. 33, 10–11 (1999)
30.M. Roessle, E. Manakova, J. Holzinger, K. Vanatalu, R.P. May, H. Heumann, Physica B 276–278, 532–533 (2000)
31.Th. Nawroth, M. Rusp, R.P. May, Physica B 350, 635–638 (2004)
32.L. Ditzel, J. Lowe, D. Stock, K.O. Stetter, H. Huber, R. Huber, S. Steinbacher, Cell 93, 125–138 (1998)
33.I. Gutsche, J. Holzinger, M. Roessle, H. Heumann, W. Baumeister, R.P. May, Curr. Biol. 10, 405 (2000)
34.I. Gutsche, O. Mihalache, W. Baumeister, J. Mol. Biol. 300, 187 (2000)
35.I. Gutsche, O. Mihalache, R. Hegerl, D. Typke, W. Baumeister, FEBS Lett. 477, 278 (2000)
36.H. Gutfreund, Kinetics for the Life Science (Cambridge University Press, University of Bristol, 1995)
37.O. Llorca, S. Marco, J.L. Carrascosa, J.M. Valpuesta, FEBS Lett. 405, 195–199 (1997)
38.A. Richardson, S.M. van der Vies, F. Keppel, A. Taher, S.J. Landry, C. Georgopoulos, J. Biol. Chem. 274, 52–58 (1999)
39.J.F. Hunt, S.M. van der Vies, L. Henry, J. Deisenhofer, Cell 90, 361–371 (1997)
40.J.F. Hunt, A.J. Weaver, S.J. Landry, L. Gierasch, J. Deisenhofer, Nature 379, 37–45 (1996)
41.J. Holzinger, Untersuchung der Reaktionszyklen von Chaperoninen aus Escherichia coli und Thermoplasma acidophilum mit Hilfe der Neutronenkleinwinkelstreuung. (PhD Thesis, LMU, M¨unchen, 2002)
42.M.B. Kozin, D.I. Svergun, J. Appl. Cryst. 34, 33 (2001)

11
Complex Biological Structures:
Collagen and Bone
P. Fratzl, O. Paris
11.1 Introduction
Biological materials such as wood, bone, or tendon are hierarchically structured and extremely complex. Properties have been shown to depend on a large extend on all size levels. For a better understanding of structure–function relations it is, therefore, necessary to get a better insight into their structure at all levels. The molecular and supramolecular levels are, in principle, accessible to electron microscopic or scattering techniques. However, the progress was slow mostly because of the di culty to disentangle the contributions of the various size levels. One of the advantages of neutron and X-ray di raction (or scattering) is the fact that information can be collected at the nanometer level in mostly intact macroscopic specimens. This advantage has been used in recent years to advance considerably the understanding of structure–function relations in collagen and bone. There are also some specific aspects where the use of (synchrotron radiation) X-rays was superior and others were the application of neutrons proved advantageous. These aspects will be worked out in more detail in the following sections. Roughly speaking, the high brilliance of synchrotron radiation is the key for position-resolved and/or time-resolved experiments. Position resolution is essential if one aims at exploring quantitatively the hierarchical nature of the structure (scanning X-ray di raction or scanning small-angle scattering). Time resolution allows to focus on mechanisms of deformation, for example, because the material structure can be monitored during the deformation. Neutrons, on the other hand, profit from the fact that the scattering length densities are not directly linked to electron densities. In particular, for organic/inorganic mixtures such as bone, information can be collected on the structure of the organic component, even in fully mineralized tissue. With X-rays on the contrary, the huge signal from the inorganic phase can completely cover the scattering contribution from the organic component. Another strength of neutron scattering in this context is the possibility of contrast variation with D2O/H2O mixtures, which allows to highlight specific components in the structure, such as covalent cross-links in
206 P. Fratzl et al.
collagen for example. Inelastic (or quasielastic) neutron scattering can also give information on protein dynamics in such systems or can be used as a spectroscopic tool to identify phase compositions. Of course, there are further applications of neutrons not related to scattering, such as neutron activation analysis of bone to study the calcium–phosphorus ratio [1]. In this chapter, we will restrict ourselves to the applications of scattering (or di raction) of neutrons and X-rays to study the complex biological materials, collagen and bone. Progress in this field has been closely linked also to the combined use of X-rays and neutrons which clearly emphasizes the complementarities of these two scattering approaches.
11.2 Collagenous Connective Tissue
11.2.1 Structure and Dynamics by Neutron Scattering
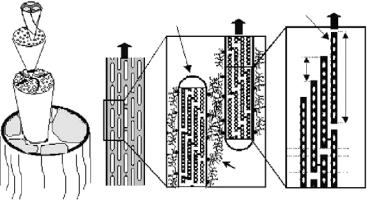
Collagen is among the most abundant proteins in vertebrates and a major constituent in many hierarchically structured biological tissues. It is found in tendon, bone, cartilage, cornea, skin, blood vessels, and other connective tissue and its function is mainly mechanical. A typical example is the tendon consisting mostly of collagen type I shown schematically in Fig. 11.1. Collagen molecules are triple helices with a length of about 300 nm. Two chains in this triplet (called type 1) are associated with a third chain (called type 2), which is similar but not identical. Molecules are assembled into fibrils by an axial staggering with a periodicity of 67 nm (Fig. 11.1d). Fibrils typically have a diameter of a few hundred nanometers. They are decorated with proteoglycans, which form a matrix between fibrils (Fig. 11.1b,c). Fibrils are assembled into fascicles and finally, into a tendon (Fig. 11.1a). While the axial stagger of the molecules in the fibrils (Fig. 11.1d) is well established, the full threedimensional arrangement is still a matter of debate. There is a predominant liquidlike order with some degree of long-range molecular ordering on a quasihexagonal lattice in the cross-section of fibrils [2,3]. The outstanding mechanical properties of tendons are due to the optimization of their structure (see Fig. 11.1) on many level of hierarchy. One of the challenges is to work out the respective influence of these di erent levels. Only a few covalent cross-links are connecting each of the collagen molecules to its neighbors [4]. Nevertheless, these cross-links are crucial for the mechanical functioning of the collagenous tissue. Indeed, when the cross-link formation is inhibited, the strength of the tissue reduces dramatically and the mechanical behavior resembles more a viscous fluid than a solid [3]. One of the strengths of neutron scattering is that – using the methodology of contrast variation by D2O/H2O substitutions – the position of cross-links may be studied within the collagen tissue. Using neutron scattering, Wess and co-workers determined two positions of natural crosslinks within the axial period of the collagen fibrils structure [5,6]. In addition, pathological cross-linking by glycation may occur in diseases such as diabetes.
|
|
11 |
Complex Biological Structures |
207 |
|
Diameter of |
|
|
|
|
|
Collagen molecule |
|
|
|
M |
eM |
1.3 nm |
M |
|
F |
eF |
|
eT |
|
||||
Collagen fibril |
|
|
|
|
|
F |
|
|
nm |
nm |
|
50− 500 nm |
|
|
|||
|
|
|
~300 |
||
Fascicle |
|
|
|
67 |
|
|
|
|
|
|
|
50− 300 μm |
|
|
|
|
|
Tendon fiber |
|
|
|
|
|
100− 500 μm |
|
|
|
|
G |
|
|
|
|
|
|
|
|
|
|
pg |
O |
|
|
|
|
|
|
|
(a) |
(b) |
(c) |
(d) |
|
Fig. 11.1. (a) Simplified tendon structure (from [3]). Tendon is made of a number of parallel fascicles containing collagen fibrils (marked F), which are assemblies of parallel molecules (marked M). (b) The tendon fascicle can be viewed as a composite of collagen fibrils (having a thickness of several hundred nanometres and a length in the order of 10 m) in a proteoglycan-rich matrix. (c) Proteoglycans are coating the fibril surfaces and connecting adjacent fibrils (F). (d) Triple-helical collagen molecules (M) are packed within fibrils in a staggered way with an axial spacing of D = 67 nm. Since the length of the molecules (300 nm) is not an integer multiple of the staggering period, there is a succession of gap (G) and overlap (O) zones. The lateral spacing of the molecules is around 1.5 nm. The full three-dimensional arrangement is not yet fully clarified, but contains both elements of crystalline order and of disorder [2]
X-ray di raction reveals that the electron density distribution along the period of the collagen fibrils changes in a systematic way with glycation [7]. Using neutron scattering and contrast variation, the glycation could be studied in even greater detail [8], giving additional information on the density and position of the cross-links along the period of the collagen fibril.
Collagen is not only the major constituent of tendon, but also of the cornea. Cornea is a tissue where collagen fibrils of uniform diameter are regularly arranged within a matrix of proteoglycans. In contrast to tendon, the fiber direction is not uniform, but the cornea consists of a succession of superposed layers with di erent fiber orientation each. The collagen fibrils in each individual layer are parallel, but not touching each other laterally as in tendon. They are kept at a significant distance from each other (probably by a thick proteoglycan layer [9]), which is essential for the transparency of the tissue (for reviews, see [10, 11]). While most structural information about cornea stems from electron microscopy and X-ray scattering, some details of the collagen structure in cornea could be better addressed by neutron rather than
208 P. Fratzl et al.
X-ray scattering [12,13]. More recently, the attachment of chlorine ions to the cornea was studied by neutron scattering and contrast variation [14].
Inelastic and quasielastic neutron scattering can generally be used to study the dynamics of crystals and molecules giving information on vibrations and on di usion. Quasielastic scattering makes generally use of the incoherent scattering cross-section which is particularly large for the hydrogen atom. Hence, in the case of collagen, the quasielastic spectrum is dominated by the protons in the structure which comprise those associated to water but also to the protein itself. First evidence of proton-dominated vibrations from collagen were reported in [15]. The dynamics of the protein associated water was studied later in more detail by quasielastic neutron scattering [16, 17]. However, due the complexity of the system with many di erent vibration modes superimposing in the neutron di raction spectrum, it was not possible until now to reach a quantitative description of vibrational modes in the protein itself.
11.2.2 Elastic and Visco-elastic Behavior of Collagen from In situ Mechanical Experiments
with Synchrotron Radiation
The outstanding mechanical properties of collagenous tissues are directly related to their hierarchical structure (Fig. 11.1). For instance, the stress– strain curve of tendon collagen shows a region of low sti ness at small strains (toe region), followed by an upward bending (sti ening) of the curve (heel region) and finally a linear region with high elastic modulus at large strains [18]. It is generally agreed that these di erent regions in the stress–strain curve are due to structural changes at the di erent hierarchical levels (Fig. 11.1), and X-ray di raction using synchrotron radiation and simultaneous tensile testing has revealed di erent strains at di erent levels in the same tendon [19–24]. The toe region can be attributed to the removal of a macroscopic crimp with a period of roughly 100 m, being visible also in the light microscope [25]. In the heel region, an entropic mechanism has been deduced from in situ synchrotron radiation studies of the equatorial scattering, which explains the increasing sti ness by straightening out molecular kinks in the gap region of collagen fibrils [20]. The linear region is thought to be due to the stretching of the collagen molecules themselves, as well as the cross-links between the molecules (Fig. 11.1), implying also a side by side gliding of the molecules [21–27]. The unique quality of such in situ experiments lies in the possibility to measure strains at di erent hierarchical levels such as the macroscopic strain of the whole tendon ( T), the strain of the collagen fibrils ( F) and the strain of the collagen molecules ( M). The finding that M < F < T underlines the importance of the cross-links as well as the influence of the proteoglycan matrix, which contributes presumably to the total tendon extension through a shear deformation. This could very recently be demonstrated by systematically investigating the strain rate dependence from tendons with normal and
11 Complex Biological Structures |
209 |
cross-link deficient collagen and a simple visco-elastic model was set up to qualitatively describe the observed e ects [24]. In this respect, we also see a great potential for in situ neutron di raction as a complementary tool to in situ X-ray studies. In situ tensile testing in di erent D2O/H2O solutions could further help to clarify the role of cross-links for the mechanical properties of tendon collagen.
11.3 Bone and other Calcified Tissue
11.3.1 Structure of Mineralized Collagen: Contributions from Neutron Scattering
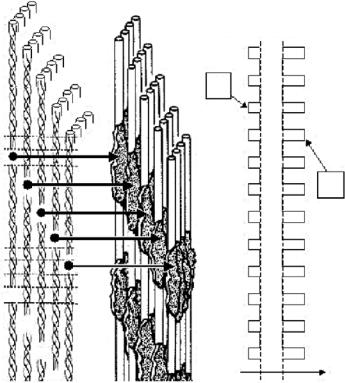
Bone consists mainly of collagen type I and mineral particles deposited inside this organic matrix. The mineral is carbonated hydroxyapatite (dahlite) in the form of extremely small particles (about 2–4 nm thickness [28, 29]) containing many crystal defects. The basic building block of bone is the mineralized collagen fibril as shown in Fig. 11.2. The collagen fibrils are similar to those in tendon (except for a di erent cross-link pattern [4]) and show a succession of gap and overlap zones along the axial direction of the fibril (see Fig. 11.2). It is believed that the mineral particles nucleate inside the gap zones [30] and then grow also into the overlap regions.
The earliest evidence for this picture of the mineralized collagen fibril came from neutron scattering investigations [31–33]. As shown in Fig. 11.2, the fact that mineral is located primarily in the gap regions changes the electron density distribution along the fibril axis from a situation where the electron density is lower in the gap (U) to one where it is higher (M). In both cases, one expects Bragg reflections along the meridional direction and they are, indeed, observed for both mineralized and un-mineralized collagen by small-angle X-ray di raction (see, e.g., [34]). When neutron are used, however, the scattering length density of the mineral is not totally dominating the signal from the protein and – using contrast variation principles – it could actually be proven that mineral particles are deposited with a periodicity corresponding to the gap-overlap succession [31, 33]. This general picture was later confirmed by transmission electron microscopy and three-dimensional reconstruction [28].
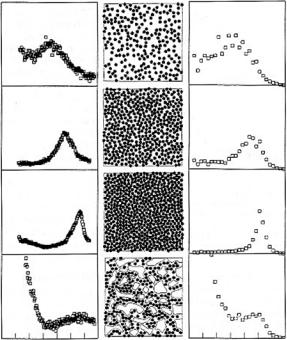
In addition to the axial structure, neutron scattering has also been operational to determine the meridional packing of collagen molecules in fully mineralized tissues [35–41]. In fact, the precise packing of collagen molecules in the fibrils has not yet been fully clarified [2,3]. Most probably, it consists of regions of three-dimensional order [42] and others with more liquid-crystalline character. A possible arrangement of molecules reconciling these aspects of order and disorder [43] is shown in Fig. 11.3. Obviously, there is a typical spacing between neighboring molecules which gives rise to a broad peak in the X-ray or neutron di raction pattern (Fig. 11.4). In this respect, it is very advantageous to use neutrons, when fully mineralized collagen tissues are to be investigated,
210 P. Fratzl et al.
|
|
electron density |
|
|
|
O |
|
|
U |
G |
|
|
O |
|
|
|
|
|
|
|
|
G |
|
|
|
O |
|
O |
|
G |
|
|
O |
|
|
G |
|
|
|
|
G |
|
|
|
|
|
|
|
|
O |
M |
|
|
G |
|
|
|
|
|
|
|
O |
|
|
|
G |
|
|
|
O |
|
O |
|
G |
|
|
O |
|
|
G |
|
|
|
|
G |
|
|
O |
|
|
|
|
O |
|
|
G |
|
|
|
|
G |
|
|
|
|
|
|
|
|
O |
|
|
|
G |
|
U |
M |
0 0 |
Fig. 11.2. Sketch of the location of mineral particles inside a collagen fibril. The un-mineralized fibril (U) is similar to the collagen fibrils found in tendon (except the covalent cross-links between the molecules which might be di erent [4]), see Fig. 11.1. Due to the axial stagger of the molecules there is a succession of gap (G) and overlap (O) zones. Mineral is thought to nucleate in the gaps and the plate-like crystals then spread into the collagen matrix. As a consequence, mineral particles are disposed according to the succession of gap and overlap zones inside the mineralized fibril (M). The drawing of mineral particles in collagen was adapted from Landis [30]. As a consequence, the axial trace of projected electron densities (right) is roughly periodic for both the un-mineralized (U) and the mineralized (M) tendon: For U the density is smaller in the gap regions and for M it is larger
since the (comparatively weak) signal from the collagen molecules can still be seen on top of the small-angle scattering from the mineral. When X-rays are used, the large electron density from the mineral can completely dominate the signal. The intermolecular spacing is in the order of 1.1 nm for fully dry fibrils. In un-mineralized collagen, the intermolecular distance can grow up to 1.6 nm and the di raction peak shifts accordingly (see Fig. 11.4). In mineralized tis-

11 Complex Biological Structures |
211 |
Fig. 11.3. Model for the cross-section through a collagen fibril according to [43]. Each dot corresponds to a collagen molecule. The lighter dots are molecules with the same axial stagger position. The darker spots are molecules with any of the other four axial stagger positions. The structure is dominated by short-range ordering (as in a two-dimensional liquid) with some elements of long-range ordering, possibly such as in grains of a polycrystal
sues, neutron scattering has shown that the swelling is limited by the amount of mineral in the fibril (see Fig. 11.5). This kind of data was essential to prove that the mineralization process of the bone matrix corresponds essentially to a replacement of water by mineral [39,40]. Therefore, the largest possible mineral contents seems to be defined by the original water content of the tissue (Fig. 11.5). Moreover, these data also show that the collagen molecules are not enclosed individually inside a mineral “wall”. They are being compressed by extra-fibrillar or interfibrillar mineral, perhaps in the way sketched in Fig. 11.4 (line IV).
Quite recently, there is a renewed interest in the actual composition of the bone mineral, which is close to, but not quite hydroxyapatite (Ca10(PO4)6OH). It is well known that the mineral is deficient in phosphate which is partially substituted by carbonate. The availability of very large neutron-scattering intensities from the nuclear-spin incoherence in hydrogen has permitted the determination of atomic motion of hydrogen in synthetic hydroxyapatite and in de-proteinated isolated apatite crystals of bovine and rat hone without the interference of vibrational modes from other structural units [44]. From such studies it was claimed initially that virtually all OH groups are also substituted by carbonate in bone mineral [44]. More recent data suggest, however, that this substitution is not complete and that at least 50% of the OH are present in bone mineral [45, 46].
212 P. Fratzl et al.
(a) |
(b) |
(c) |
I
II
III
IV
0 |
5 nm−1 |
0 |
5 nm−1 |
Fig. 11.4. (a) Meridional X-ray di raction peak as a function of the length of the scattering vector q from turkey leg tendon with di erent degrees of hydration from fully wet (I) to dry (III). The peak is seen to shift to larger q-values indicating that molecules come closer with drying. (b) shows a simulated arrangement of molecules with a calculated di raction pattern as shown in (c). The last line (IV) is a proposed arrangement for mineral particles in bone. The data (IVa) are from the mineralized part of the turkey leg tendon (figure taken from [34])
11.3.2 Investigating the Hierarchical Structure of Bone
Bone is a hierarchical tissue which adapts its detailed structure to the functional needs [47]. As an example, Fig. 11.6 shows several hierarchical levels in the shaft of a typical mammalian long bone (femur, tibia, etc.). The outer shells of such bones are made of compact bone which has typically a lamellar character (Fig. 11.6b). The lamellae consist of layers of parallel mineralized collagen fibrils (Fig. 11.6c). The typical diameter of collagen fibrils is in the range of a few hundred nanometers and they are reinforced by platelets with a thickness of 2–4 nm. These fibrils are arranged in parallel to form layers, whose orientations turn in a plywood-like manner [48, 49]. Since the mineral particles are crystalline, their Bragg reflections can be used to determine the crystallographic orientation distribution of the mineral particles in the entire