

Neutron Scattering in Biology - Fitter Gutberlet and Katsaras
.pdf80 P. Timmins
occupied by two detergent/amphiphile micelles which must have displaced the original lipid during purification.
5.4.2 Porins
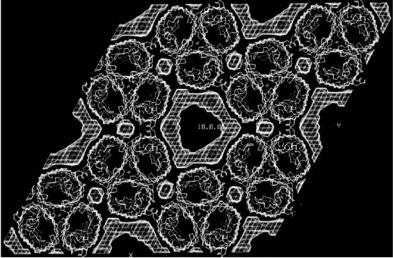
Another class of membrane proteins to be studied were the outer membrane proteins known as porins. In contrast to the bacterial photosynthetic reaction centers these proteins form multistranded β-barrel structures which are deeply embedded in the membrane with relatively small protruding loops. The OmpF porin from E. coli was in fact, along with the photosynthetic reaction center from R. viridis, the first membrane protein to be crystallized in the early 1980s [19, 20]. Due however to technical crystallographic problems its structure was not solved until 1994 when a trigonal form was crystallized and served as model for a molecular replacement solution [21]. The neutron crystallographic analysis of the tetragonal form [22] showed very clearly the detergent belt bound to the hydrophobic surface of the protein and suggested how the protein is anchored in the membrane. Figure 5.4 shows how the detergent binding surface is clearly delimited by two lines of aromatic amino acids with tyrosine residues directed towards the headgroups and phenyl alanines towards the hydrophobic acyl chains. This had been surmised from the X-ray structure but the absence of any density in the electron density maps did not allow this to be demonstrated. Another important observation to come from the neutron di raction analysis concerned the packing of the molecules
Fig. 5.4. Detergent binding in tetragonal crystals of OmpF porin. Note the binding surface delimited by aromatic residues
5 Membrane Protein Crystals by Neutron Crystallography |
81 |
in the crystal. The X-ray structure shows the OmpF trimers to pack as two interpenetrating lattices with no protein–protein contacts between molecules in the separate lattices. The question then arises as to how such a structure is stabilized. The answer was provided by the neutron data which showed the two lattices to come into contact through the detergent belts of adjacent molecules. The crystal is therefore stabilized by protein–protein, protein–detergent, and detergent–detergent contacts.
The trigonal form of OmpF is very di erent from this [23]. The OmpF trimers here form columns of molecules running in opposite directions with protein–protein contacts being responsible for the interactions within each column. The columns of molecules are then held together by fusion of the detergent rings surrounding the hydrophobic protein surfaces. No distinct detergent belts remain but protein trimers interact directly through hydrophobic interactions or mediated by patches of detergent (Fig. 5.5). X-ray contrast variation using xenon has also been used to investigate detergent binding in these crystals [24]. Although the detergent containing volumes determined by neutrons and X-rays partly overlap there are significant di erences. This is due most probably to the xenon penetrating only the most hydrophobic parts of the detergent (the tails) as well as into the surface of the protein [24].
Another outer membrane protein to have been studied is the outer membrane phospholipase A (OMPLA) from E. coli. The structure of this protein is a 12 stranded β-barrel and its active form is a dimer [25]. Here the neutron data showed a detergent structure distinct from any other of the known
Fig. 5.5. Neutron scattering density map showing the detergent distribution (hashed regions) in trigonal crystals of OmpF porin from E. coli. Note the hydrophobic protein–protein contacts
82 P. Timmins
structures with a continuous detergent phases throughout the crystal, somewhat reminiscent of lipid cubic phases [26]. An attempt was also made in this case to obtain the structure from X-ray di raction of Xe soaked crystals but the results were not consistent with the neutron maps.
5.5 Conclusions
Neutron low resolution crystallography is a powerful technique for visualizing disordered regions in crystals of biomolecular complexes and in particular the localization of detergent in crystals of membrane proteins. The technique of course relies on the availability of crystals but these do not necessarily have to be very large as required for high resolution crystallography −0.1 mm3 or less is su cient. The membrane protein structures studied to date have demonstrated that not only are protein–protein interactions important in the formation of crystals but that in some cases protein–detergent and even detergent–detergent interactions can be crucial.
References
1.W.L. Bragg, M.F. Perutz, Acta Cryst. 5, 277 (1952)
2.W. Shepard, R. Kahn, M. Ramin, R. Fourme, Acta Cryst. D56, 1288–1303 (2000)
3.H. Tsuruta, Synchrotron Rad. News 13, 10 (2000)
4.M. Roth, A. Lewit-Bentley, Acta Cryst. A38, 670 (1982)
5.M. Roth, A. Lewit-Bentley, G.A. Bentley, in Position Sensitive Detection of Thermal Neutrons, P. Convert, J.P. Forsyth (Eds.) (Academic Press, New York, 1983)
6.S. Penel, P. Legrand, J. Appl. Cryst. 30, 206 (1997)
7.W. Kabsch, J. Appl. Cryst. 26, 795 (1993)
8.M. Roth, A. Lewit-Bentley, G.A. Bentley, J. Appl. Cryst. 17, 77 (1984)
9.M. Roth, Acta Cryst. A42, 230 (1986)
10.M. Roth, Acta Cryst. A43, 780 (1987)
11.T.L. Blundell, L.N. Johnson, Protein Crystallography (Academic Press, London, 1976)
12.M. Roth, in Crystallographic Computing 5, D. Moras, A.D. Podjarny, J.C. Thierry (Eds.) (Oxford University Press, Oxford, 1992)
13.M. Roth, B. Arnoux, A. Ducruix, F. Reiss-Husson, Biochemistry 30, 9403 (1991)
14.M. Roth, A. Lewit-Bentley, H. Michel, J. Deisenhofer, R. Huber, D.Oesterhelt, Nature 340, 659 (1989)
15.H. Michel, J. Mol. Biol. 158, 567 (1982)
16.P. A. Timmins, J. Hauk, T. Wacker, W. Welte, FEBS Lett. 280, 115 (1991)
17.P. Gast, Hemelrijk, A.J. Ho , FEBS Lett. 337, 39 (1994)
18.S.M. Prince, T.D. Howard, D.A.A. Myles, C. Wilkinson, M.Z. Papiz, A.A. Freer, R.J. Cogdell, N.W. Isaacs, J. Mol. Biol. 326, 307 (2003)
19.R.M. Garavito, J.P. Rosenbusch, J. Cell. Biol. 86, 327 (1980)
5 Membrane Protein Crystals by Neutron Crystallography |
83 |
20.R.M. Garavito, J.A. Jenkins, J.M. Neuhaus, A.P. Pugsley, J.P. Rosenbusch, Ann. Microbiol. (Paris) 133A, 37 (1982)
21.S.W. Cowan, T. Schirmer, G. Rummel, M. Steiert, R. Ghosh, R.A. Pauptit, J.N. Jansonius, J.P. Rosenbusch, Nature 358, 727 (1992)
22.E. Pebay-Peyroula, R.M. Garavito, J.P. Rosenbusch, M. Zulauf, P.A. Timmins, Structure 3, 1051 (1995)
23.S. Penel, E. Pebay-Peyroula, J.Rosenbusch, G. Rummel, T. Schirmer, P.A. Timmins, Biochimie 80, 543 (1998)
24.O. Sauer, M. Roth, T. Schirmer, G. Rummel, C. Kratky, Acta Cryst. D58, 60 (2002)
25.H.J. Snijder, I. Ubarretxena-Belandia, M. Blaauw, K.H. Kalk, H. Verheij, M.R. Egmond, N. Dekker, B.W. Dijkstra, Nature 401, 717 (1999)
26.H.J. Snijder, P.A. Timmins, K.H. Kalk, B.W. Dijkstra, J. Struct. Biol. 141, 122 (2003)

6
High-Angle Neutron Fiber Di raction
in the Study of Biological Systems
V.T. Forsyth, I.M. Parrot
6.1 Introduction
Fiber di raction has made a critical impact in structural biology. It has broad application to the study of a wide range of biological and synthetic polymers, and continues to provide key information about the structure of these molecules. The purpose of this chapter is to illustrate the general scope of the method and in particular to demonstrate the impact of neutron fiber di raction methods for the study of biological systems. Because in most neutron fiber di raction work is carried out in combination with X-ray di raction studies, this review will focus on the unique complementarity provided by the two approaches to structure analysis. Two specific examples have been chosen to illustrate the power of a combined X-ray and neutron approach by high-angle fiber di raction – DNA, and cellulose. Both are examples where the involvement of neutron studies has added completely unique information.
The expanding interest in structural studies of biological and industrial polymers is opening up important new opportunities for neutron fiber diffraction. This is illustrated by current trends in X-ray fiber di raction methods where there is increasing emphasis on the exploitation of the high fluxes available for time-resolved studies and for microdi raction work, and the key aspects of hydration and hydrogen bonding, both of which are largely inaccessible to X-ray fiber di raction methods, but which nonetheless having a vital bearing on biological function or physical properties of biological and synthetic polymers. Many of the assumptions made on the basis of initial X-ray fiber di raction analyzes, even for relatively simple systems, are being reevaluated through the use of innovative techniques at modern X-ray and neutron sources. This is likely to become more important in the future as the new instruments and facilities currently being constructed make their impact.
86 V.T. Forsyth et al.
6.2 Fibers and Fiber Di raction
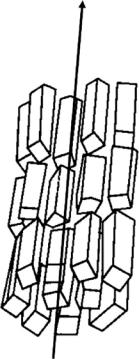
An aligned fiber is characterized as containing many regions in which the polymer molecules are arranged with their long axes parallel to the fiber axis. These are typically separated by less ordered amorphous regions. The molecular alignment within such a fiber is associated with varying degrees of order in the side-by-side packing of the chains [1–3]. To a large extent this ordering will determine the general appearance of the observed diffraction pattern which may vary from continuous molecular transform to the other extreme where the pattern is dominated by sharp Bragg reflections. Because there is no preferred orientation of microcrystallites about the fiber axis, an X-ray fiber di raction pattern from a crystalline fiber has some of the character of that which would be obtained from a single crystal if it was rotated by 360◦ about one of its principal axes during data collection (see Fig. 6.1). The resolution attained in fiber di raction experiments varies widely but can in favorable circumstances extend to atomic resolution. While there is some loss of information imposed by cylindrical averaging in fiber di raction this has to be o set against the fact that in
Fibre axis
Fig. 6.1. Diagram to illustrate the random orientation of crystallites about the fiber axis in a crystalline fiber
6 High-Angle Neutron Fiber Di raction |
87 |
most situations comparable measurements are not possible from single crystal systems.
In many biological structures such as muscle and connective tissue and also in synthetic industrial polymers, the fibrous state is functionally important and fiber di raction provides the most powerful technique to establish both molecular structure and the higher order interactions of these molecules. It is also true to say that fiber di raction o ers one of the few quantitative methods which are well suited to the study of conformational pathways in polymer molecules. The fibrous state, with its typical mix of highly ordered and amorphous regions, can accommodate changes in molecular conformation and packing much more readily than can single crystals. This capability allows a systematic investigation of the dependence of molecular conformation on important biological parameters such as water and ionic contents in a fiber. In particular, conditions can be chosen which are much closer to the in vivo environment than is typically achieved in solution or in single crystal studies. In many biological systems changes in the water content of a fiber can be controlled by varying the relative humidity of the fiber environment, and it is possible to use synchrotron X-ray sources to carry out time-resolved studies of stereochemical pathways followed in structural transitions, that occur as a function of hydration. While this type of work provides quite novel information through the capacity to carry out time-resolved [4, 5] and even microdi raction studies [6], X-ray di raction is often a poor probe of water structure. It is fortunate therefore that neutron di raction can be used, particularly in conjunction with hydrogen/deuterium isotopic replacement, to study the location of water or hydrogen in biopolymer molecules.
6.3 Neutron Fiber Di raction: General Issues
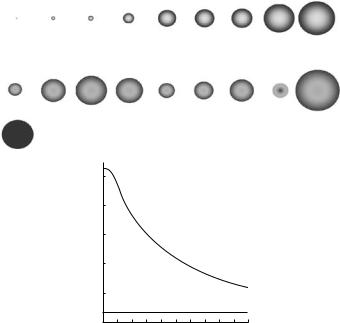
The advantages of neutron di raction are well known. One of the most important of these is the fact that, in contrast to the situation with X-rays, hydrogen atoms have a coherent scattering length that is comparable to the other atoms commonly found in biological systems (Fig. 6.2).
Furthermore, it is relatively easy to exchange hydrogen by its more strongly scattering isotope, deuterium, as illustrated in Fig. 6.3, and to use this isotopic replacement in determining the location of hydrogen/water in these systems. Although it has been argued (for the single crystal case) that this type of information can be obtained from high resolution (i.e., atomic) X-ray di raction studies, the real situation is more complex. Not only is it quite rare (especially in fibrous systems) for biological samples to di ract to atomic resolution, but there is evidence that even when they do, hydrogen atoms with large thermal displacement parameters are not well located. Neutron methods identify such atoms, which may be of biological interest, very clearly [7], even when the available resolution is relatively low.
88 V.T. Forsyth et al.
X-ray
Scattering proportional to Z
H |
B |
C |
O |
Al |
Si |
P |
Ti |
Fe |
1 |
3 |
4 |
8 |
13 |
14 |
15 |
22 |
26 |
Neutron
Scattering not proportional to Z
−3.74 (H) 5.30 |
6.65 |
6.67 (D)
|
5 |
|
amplitude |
4 |
|
3 |
||
Scattering |
||
1 |
||
|
2 |
5.80 3.45 4.15 5.13 −3.44 9.45
X-rays
Neutrons
0.2 |
0.4 |
0.6 |
0.8 |
1.0 |
|
(sin q)/l |
|
|
|
Fig. 6.2. Top: diagram giving a representation of the neutron scattering lengths, including some atoms of relevance to biological systems, and emphasizing the di erence in neutron scattering lengths of H and D. Bottom: plot showing the variation of atomic form factor for X-ray and neutron scattering as a function of scattering angle
Another important aspect of neutron scattering arises from the fact that the atomic nucleus from which neutrons are scattered is essentially pointlike in size compared to the wavelength of the incident beam. As a result, in contrast to the situation for X-rays, there is e ectively no variation in the atomic form factor for neutrons over the relevant range of scattering angle. This means that in circumstances where the observed di raction is not dominated by either thermal or spatial disorder, there exists the possibility to record neutron di raction data at higher resolution than would be possible with X-rays. This point is well illustrated in Fig. 6.4, which compares X-ray and neutron fiber di raction patterns recorded from a non-biological fiber, poly(p-phenylene terephthalamide) (PPTA) more commonly known as the commercial fibres Kevlar or Twaron.
6 High-Angle Neutron Fiber Di raction |
89 |
(a) |
(b) |
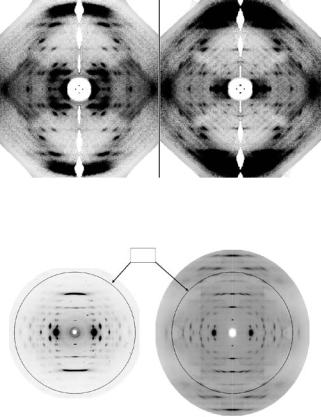
Fig. 6.3. Neutron fiber di raction patterns recorded from DNA. (a) the pattern recorded when the DNA is hydrated with D2O; (b) the pattern recorded when the DNA is hydrated with H2O
1.4A
Fig. 6.4. X-ray and neutron fiber di raction patterns recorded from a synthetic crystalline system, illustrating a fallo in the overall intensity distribution at high angles in the case of the X-ray pattern. In contrast, scattering of similar average intensity is observed well beyond this point in the neutron di raction pattern
Because of the lower brilliance of the neutron beams, there are major issues to be faced for neutron di raction studies of biological systems. By far the most important is the requirement for large sample size. Although this is a major obstacle in crystallography, there are various approaches that can be adopted in tackling this problem for fiber studies. The first high-angle studies of DNA at the ILL [8–10] were carried out using individual fibers (each about 100 m in diameter) aligned in a parallel array. In later work, sheet samples made using the wet spinning method of Rupprecht [11] were used to obtain the required sample volume both for fiber di raction and for neutron and X-ray spectroscopy [12,13]. For the work on cellulose described in the following sections of this chapter, Nishiyama and colleagues have used a shear flow method to deposit aligned microcrystals in a gel-like film which is then dried
90 V.T. Forsyth et al.
to produce large high-quality samples of various forms of cellulose that di ract to nearly atomic resolution.
For crystallographic and fiber studies in particular, the issue of sample size is further exacerbated by incoherent scattering from hydrogen in the solvent and in the macromolecule itself. It is relatively straightforward to replace H2O solvent by D2O for these studies; this results in a huge decrease in the hydrogen incoherent scattering “background”, as illustrated in Fig. 6.5.
However, there is still a large problem arising from the hydrogen that is covalently bound to carbon atoms in the structure. This places limitations on sample size, data collection times, on the interpretation of the data, and indeed on the quality of the final analysis. However, the presence of hydrogen incoherent scattering and the associated limitations in neutron scattering can be e ectively eliminated by sample perdeuteration. Although the benefits of sample deuteration have been widely appreciated for some time, it has never been easy for individual biologists to deuterate their systems in a routine way that allows best use to be made of valuable central facility resources; the expertise is rather and individual requirements vary quite considerably. As described in the concluding sections of this chapter this problem has been identified at the ILL and the EMBL in Grenoble [14] where these facilities, with advice from their user communities and a number of peer-review committees, took the view that major developments for modern instrumentation should be paralleled with strong emphasis on sample preparation issues. Of particular importance in this area is the utilization of selective and sample deuteration.
6.4 Facilities for Neutron Fiber Di raction
Neutron high-angle fiber di raction methods were developed at the ILL on the D19 di ractometer, and most of the work that has been published subsequently has used this facility. D19 was originally optimized for chemical crystallography where it was used for structure determination for systems having relatively large unit cell dimensions [15–19]. A key aspect of the instrument was the use of an area detector rather than smaller “single” detectors commonly used in crystallographic studies of small molecule systems. Such a detector is essential for fiber di raction work, which requires not only that the intensities of the Bragg reflections are well measured, but also that the variation of the background over the entire pattern is measured as accurately as possible. Figure 6.6 shows a photograph of the D19 di ractometer alongside a sample dataset.
At the time that this photograph was taken the D19 detector was a long thin banana-shaped gas-filled device with an angular aperture of 4◦ in its equatorial plane and 64◦ in its vertical plane. The requirements for data collection, data reduction and analysis are di erent from those commonly used in single-crystal studies. The strategy for data collection has therefore always