

2.8 Равновесие в гетерогенных системах, состоящих
из двух и более компонентов
2.8.1 Равновесие жидкость – пар. Закон Рауля
Пусть в состоянии равновесия находятся жидкость и выделяющийся из нее пар. В соответствии с теоремой равновесия Гиббса химический потенциал любого компонента в системе одинаков во всех фазах:
![]() .
.
Выразим химический потенциал i-го компонента в жидкой фазе через его термодинамическую активность, а в паровой фазе – через его парциальное давление, то есть примем, что паровая фаза идеальна. Тогда получим:
![]() .
.
Преобразуем уравнение:
![]() .
.
Значение выражения,
заключенного в скобки, найдем из
граничного условия: если xi
= 1, то γi
= 1,
![]() и
и![]() ,следовательно,
,следовательно,
![]() ,
,
где
![]() – давление насыщенного пара над
индивидуальнымi-м
компонентом при температуре раствора.
– давление насыщенного пара над
индивидуальнымi-м
компонентом при температуре раствора.
Подставим в уравнение вместо выражения в скобках его значение:
![]() ,
,
![]() .
.
Выразив активность через мольную долю и коэффициент активности, получим:
![]() ,
(2.8)
,
(2.8)
Уравнение (2.8) выражает основной закон жидких растворов – закон Рауля.
Применительно к идеальным жидким растворам (γi → 1) уравнение (2.8) запишется
![]() .
.
Рассмотрим идеальный жидкий раствор, состоящий из летучих компонентов А и В, тогда:
![]()
Выразим мольную долю компонента А через мольную долю компонента В:
хА = 1 – хВ.
Общее давление над раствором складывается из парциальных давлений компонентов р = рА + рВ, тогда
![]()
Таким образом, зависимость давления насыщенного пара компонентов и общего давления пара от состава раствора является линейной.
Зависимость давления насыщенного пара от состава двухкомпонентного раствора представляется в виде диаграммы р(рА, рВ) – х (рис. 2.2).
Д ля
описания зависимости давления насыщенного
пара компонента от состава реального
раствора используют закон Рауля в виде:
ля
описания зависимости давления насыщенного
пара компонента от состава реального
раствора используют закон Рауля в виде:
![]()
Для реального раствора, содержащего летучие компоненты А и В общее давление над раствором равно:
![]() .
.
П
Рис.
2.2. Зависимость давления паров
от состава раствора
Отклонения давления пара от линейной зависимости в сторону больших значений называют положительными, а в сторону меньших значений – отрицательными. Величина и вид отклонений зависят от сил взаимодействия между частицами. Силы взаимодействия межу частицами идеального раствора такие же, как и в чистых жидкостях, поэтому отклонений не возникает. В реальных растворах при уменьшении сил межмолекулярного взаимодействия усиливается испарение. Следовательно, увеличивается давление насыщенного пара и возникает положительное отклонение от линейной зависимости. Закономерности для растворов с отрицательными отклонениями имеют противоположный характер (рис. 2.3).
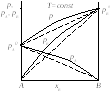

а б
Рис. 2.3. Диаграмма состояния реальных растворов: а – с отрицательными отклонениями; б – с положительными отклонениями
Состав равновесного с жидким раствором пара (уi) определяется согласно закону Дальтона:
![]() –для идеального
раствора;
–для идеального
раствора;
![]() –для реального
раствора.
–для реального
раствора.
Для представления данных по фазовому равновесию жидкость-пар кроме рассмотренных диаграмм применяются также диаграммы давление пара – состав (р – х(у)), температура кипения – состав (Т – х(у)) и состав пара – состав раствора (y – x) (рис. 2.4).
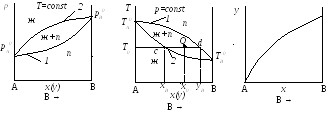
Рис. 2.4. Диаграммы давление пара – состав (р – х(у)), температура кипения –
состав (Т – х(у)) и состав пара – состав раствора (y – x)
На представленных диаграммах:
точке А соответствует чистому компоненту А (100 %);
точке В – чистому компоненту В (100 %);
На диаграмме р – х(у):
1 – кривая зависимости давления насыщенного пара от состава пара;
2 – кривая зависимости давления насыщенного пара от состава жидкости.
Область ниже кривой состава пара отвечает состоянию гомогенной системы – пара; область выше кривой состава жидкости отвечает состоянию жидкости.
На диаграмме Т – х(у):
1 – кривая зависимости температуры кипения от состава пара;
2 – кривая зависимости температуры кипения от состава жидкости.
Область выше кривой состава пара отвечает состоянию гомогенной системы – пара; область ниже кривой состава жидкости отвечает состоянию жидкости.
Область, лежащая между кривыми состава пара и состава жидкости отвечает состоянию гетерогенной системы жидкость-пар, любая точка внутри этой области характеризует состояние равновесия между жидкостью и паром. Например, на диаграмме Т – х(у) в точке О при температуре Т0 находится гетерогенная система, состоящая из жидкости и пара общий состав которой равен х0. Точки с и d выражают составы равновесных жидкости хВ и пара уВ.
Таким образом, составы жидкости и равновесного с нею пара в общем случае неодинаковы.
Если известна масса гетерогенной смеси в точке О m, то по правилу рычага можно рассчитать массы равновесных фаз:
 .
.
Разделение жидких летучих смесей.
Различие составов жидкости и равновесного с нею пара имеет большое практическое значение, так как позволяет разделять летучую смесь на практически чистые компоненты. Основными методами разделения жидких летучих смесей является перегонка и ректификация. Эти методы основаны на испарении летучей смеси и конденсации образовавшегося пара.
Различают простую и фракционную перегонку или ректификацию. При простой перегонке нагревание жидкости сопровождается непрерывным отбором пара и его конденсацией. В результате простой перегонки можно выделить практически в чистом виде только один из компонентов (менее летучий компонент). Более летучий компонент при простой перегонке в чистом виде не выделяется.
Фракционная перегонка заключается в многократном повторении процессов испарения и конденсации и позволяет разделить раствор на чистые компоненты. На производстве фракционная перегонка осуществляется непрерывно и называется ректификацией.
Основными законами перегонки и ректификации являются законы Коновалова.
Первый закон Коновалова: пар по сравнению с жидкостью обогащен тем компонентом, добавление которого к бинарной смеси увеличивает общее давление пара при данной температуре или понижает температуру кипения при данном давлении.
Из первого закона Коновалова следует, что пар по сравнению с жидкостью обогащается более летучим компонентом. Таким образом, путем многократного испарения жидкости и конденсации пара можно добиться практически полного разделения компонентов.
Р Рис.
2.4.
Компонент В является более летучим по сравнению с компонентом А, так как его температура кипения ниже. Согласно диаграмме данный раствор закипит при температуре Т и появится пар, состав которого равен уВ′. Сконденсировав образовавшийся пар, получим жидкую смесь, мольная доля компонента В в которой составит хВ′, причем хВ′ > xB. Если снова нагреть полученную смесь, то получим пар состава уВ″, при конденсации которого образуется жидкая смесь такого же состава хВ″ > хВ′. Повторяя указанные действия, получим практически чистые компоненты А и В (хВ′″ ≈ 100 %).
Для реальных растворов, характеризующихся значительными положительными или отрицательными отклонениями, компоненты которых имеют очень близкие температуры кипения или давления насыщенных паров, на кривых жидкости и пара наблюдаются точки экстремума.
Второй закон Коновалова: точкам экстремума на кривой зависимости общего давления пара от состава раствора (температуры кипения раствора от состава раствора) соответствуют азеотропные нераздельно кипящие смеси, в которых состав жидкости и состав равновесного с нею пара одинаковы.
Диаграммы состояния таких растворов представлены на рис. 2.5.
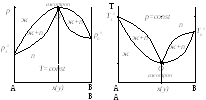
Рис. 2.5. Диаграммы р – х2(у2) и t – х2(у2) с азеотропом
в минимуме давления (максимуме температуры кипения)
Примером смеси, образующей азеотроп, является бражка, основными компонентами которой являются этиловый спирт и вода. При концентрации этилового спирта, равной 96 % об. образуется азеотропная смесь. Поэтому путем обычной ректификации получить 100 %-го этиловый спирт нельзя.
На практике для устранения азеотропа предпринимают различные меры, например, повышают внешнее давление, в результате увеличивается температура кипения смеси и азеотроп исчезает или изменяется его состав.