

Крючков Фундаменталс оф Нуцлеар Материалс Пхысицал Протецтион 2011
.pdf∙determination of the count rates nip in a series of peaks that belong to
one isotope and calculation of |
the values nip / Iγi |
that |
characterize the |
|
efficiency εγi . The |
quantities |
Iγi are known for |
each |
group of quanta |
forming these peaks |
Si; |
|
|
|
∙the obtained values εγi for a series of peaks are used to plot the
dependence of εγ on Еγ .
The biggest difficulty involved in handling the range of 84–130 keV, that is similarity of the radiations measured by energies, is coped with by using high-resolution Ge-detectors and a special spectrum decomposition
program called MGAU (Fig. 5.7).
To determine the relative content of 235U, X-ray peaks of 89.95 keV (Th XKα2) and 93.35 keV (Th XKα1) are used. For 238U, a doublet of overlapped gamma peaks of 92.37 keV and 92.79 keV (234Th) is used.
The content of 234U is found from the gamma-ray line of 121 keV. The enrichment determination error makes not more than several percent for
uranium samples in a range from depleted to highly enriched uranium (from 0.3% to 93% of 235U).
inCount pulchannel,
Energy, keV
Fig. 5.7. Uranium radiation spectrum in the energy range 88–100 keV and the outcome of its mathematical decomposition into separate components
241

Constraints: the gamma doublet of 92.37 keV and 92.79 keV used to determine the relative content of 238U belongs to 234Th, which is the product of the α-decay of 238U. Therefore, the daughter 234Th in the sample should be in equilibrium with the parent 238U, which occurs only in approximately 5 months after the chemical isolation of uranium. The container wall thickness influences the accuracy of analysis: a 16-mm wall of a steel container with UF6 attenuates the radiation of 90–100 keV by a factor of approximately 250.
Nondestructive gamma ray spectrometry measurements of plutonium isotopic composition
Most of the Pu samples contain 238Pu, 239Pu, 240Pu, 241Pu and 242Pu. Besides, these always include 241Am and 237U, which are the products of 241Pu decay. Data on the decays of the above isotopes are presented in Table 5.5.
Table 5.5
Decay data of selected isotopes
Isotope |
Т1/2, years |
Activity, |
|
decay/s×g |
|||
|
|
||
238Pu |
87.74±0.04 |
6.33×1011 |
|
239Pu |
24119±26 |
2.2942×109 |
|
240Pu |
6564±11 |
8.3971×109 |
|
241Pu |
4.348±0.022 |
3.8244×1012 |
|
242Pu |
376300±900 |
1.4522×108 |
|
241Am |
433.6±1.4 |
1.2655×1011 |
|
237U |
6.75 days |
9.4080×107 |
* Given there is equilibrium with 241Pu.
Gamma measurements determine the isotopic relations of f238/f239,
f240/f239, f241/f239, f242/f239, with the total of f238+f239+f240++f241+f242=1, where
fi is the fraction of the i-th plutonium isotope. Possible compositions of Pu isotopes are presented in Table 5.6.
If to measure the intensities of gamma radiations from the isotopes contained in a sample, self-calibration may help determine the relative efficiencies of the detection thereof. Self-calibration technique is based on using a series of peaks of one isotope contained in the analyzed sample, and on the quantum yield data.
242

|
|
|
|
|
Table 5.6 |
|
Possible isotopic compositions of plutonium |
|
|
||||
|
|
|
|
|
|
|
Isotope |
238Pu |
239Pu |
240Pu |
241Pu |
242Pu |
|
|
|
|
|
|
|
|
Low burnup, % |
0.01 |
94 |
5.5 |
0.5 |
0.03 |
|
|
|
|
|
|
|
|
High burnup, % |
2 |
57 |
25 |
11 |
5 |
|
|
|
|
|
|
|
|
The sample mass may vary from 100 mg to the limits imposed by nuclear safety and security considerations. The matrix can be any (solid, liquid, powder, heterogeneous). There are no geometry restrictions. The age from the time of the Pu chemical isolation can be also any. Both planar and coaxial Ge-detectors can be used for measurements.
There are different ranges of gamma-ray energies that contain radiations of plutonium isotopes. Within all ranges, helpful data can be found in the peaks of various isotopes which are close by energy. Measurements in highenergy regions enable the Pu contained inside thick-wall containers to be measured without having the sample taken out. This results in a higher safety of the operator and makes it possible to inspect sealed containers used for long-term NM storage.
To measure samples of a large mass, one can use radiations with the energies 642.5 keV for 240Pu, 646.0 keV for 239Pu and 766.4 keV for 238Pu,
which are highly penetrable and enter the detector from across the sample thickness.
Measurements of samples in thin-wall containers are done in lowenergy regions with the aid of planar Ge-detectors. Coaxial detectors are used both for thin-wall and thick-wall containers.
Planar detectors with an area of 200–500 mm 2 and a thickness of 10–13 mm are often used. Such dimensions offer a compromise, which is helpful in getting a good resolution and an acceptable efficiency.
Use of planar detectors is confined to the region of ≤400 keV. Measurements in higher-energy regions need a coaxial detector with a
Minimum reported quantity.
243
resolution of ≤1.7 keV for the peak of the 60Со calibration source of the energy 1332 keV.
Test measurements of plutonium samples are conducted based on MGA and FRAM codes.
MGA (multi-group analysis) is a code to determine the isotopic composition of Pu from results of gamma ray spectrometry measurements in the energy range of 99–104 keV. All intensive ga mma peaks and X-ray peaks in this region are used. No entry of data (coming from the analog-to- digital converter or ADC) or efficiency calibration is required.
The code has the following drawbacks:
∙there is a problem of identifying 242Pu that cannot be measured directly due to a very low intensity of its gamma radiation. A complete analysis of
plutonium composition requires either a destructive analysis (mass spectrometry) or estimation of the 242Pu content by isotopic correlation method;
∙it is not applicable where there is a strong background of fission fragments and to measurements of samples in thick-wall containers.
Using FRAM code requires the sample gamma radiation spectrum measured on equipment with a high energy resolution and a set of parameters to be used for the analysis. The parameters control the search for peaks and specify the analysis scope and the isotopes to be determined.
FRAM code features:
∙ease of adaptation to various measurement conditions with no need for reprogramming;
∙a user capability to edit the analysis parameters;
∙a capability of analyzing all kinds of samples having the following contents (2–38% of 240Pu, 0.01–50% of 241Am) and samples in thick-wall containers.
Table 5.7 gives data on accuracies in nondestructive measurements of plutonium isotopic compositions.
Table 5.7
Accuracy of nondestructive measurements for plutonium isotopic compositions, %
Еγ, range, keV |
Тmeas |
238Pu |
239Pu |
240Pu |
241Pu |
40–60 |
10–300 min |
0.3–5.0 |
0.05–0.5 |
0.2–1.0 |
0.2–1.0 |
90–105 |
30–60 min |
0.3–5.0 |
0.05–0.5 |
0.2–1.0 |
0.2–0.8 |
>120 |
1–4 h |
1–10 |
0.1–0.5 |
1–5 |
0.3–0.8 |
244
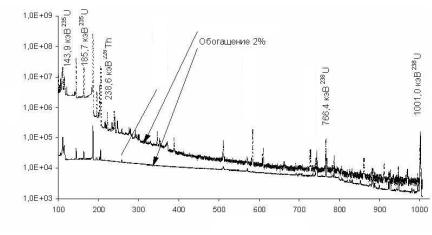
FRAM can be also used to examine uranium samples, specifically to test enrichments of uranium articles inside a container. The thing is that commonly used MGAU-based analyses rely on soft radiation measurements and are not suitable for testing shielded samples. Using FRAM enables use of harder radiations for analyses by measuring the spectrum thereof with the aid of a coaxial НРGe-detector. Fig. 5.8 presents the radiation spectra for differently enriched uranium samples obtained with the aid of a coaxial HPGe-detector.
|
U |
|
U |
|
|
|
|
235 |
|
235 |
|
|
U |
|
keV |
|
keV |
|
|
|
|
|
|
|
238 |
||
Count |
|
|
Th |
|||
143.9 |
|
185.7 |
|
keV238.6 |
258.4keV |
|
|
|
|
|
|
228 |
|
|
|
|
|
|
|
|
Enrichment 45% |
|
|
|
|
Enrichment 2% |
|
|
|
|
Th |
|
|
|
U |
Th |
U |
|
238 |
|
228 |
228 |
238 |
|
|
583.1keV |
727.3 keV |
766.4 keV |
|
1001.0keV |
|
|
|||
|
|
|
|
Energy, keV
Fig. 5.8. Spectra of uranium sample radiation
Standards have quite different roles in gamma ray spectrometry measurements of Pu (and U) isotopic compositions as compared to other nondestructive test techniques. The values sought for in gamma ray spectrometry are obtained using certain physical constants and selfcalibration results. Standards are used just to control measurement results.
Analyses of NM solutions. Densitometry
K-edge or L-edge densitometry (KED or LED) is based on measuring the transmission of a highly collimated photon or gamma-quanta beam from an external source through an NM solution sample. The technique is practically insensitive to the radiation emitted by the sample itself (for example, high content of fission products in the solution is allowable). The
245
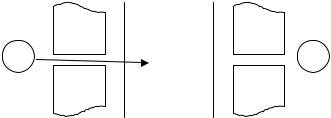
collimator confines observation to only a small sample portion. For this reason, the sample should be strictly homogeneous throughout the thickness. K-edge or L-edge densitometer is the instrument for nondestructive assays of NM concentrations in solutions (Fig. 5.9).
Detector
S  D
D
Source
Collimator |
Sample |
Collimator |
|
|
|
Fig. 5.9. Diagram of densitometry measurements
The absorption factor for each element is measured in steps with the energies being equal to the energies of the electron coupling on the K- and L- shells of atoms (Table 5.8).
Measuring the transmission of one-energy radiation through a sample may give information on the content of one material or one component of a mixture. Using a radiation of two different energies makes it possible to control the composition of a two-component mixture.
Table 5.8
Energy of electron coupling on K- and L-shells in various NM, keV
Element |
Th |
U |
Np |
Pu |
Am |
K-edge |
109.56 |
115.61 |
118.68 |
121.82 |
125.03 |
L-edge |
16.3 |
17.2 |
17.6 |
18.0 |
18.5 |
A measurement of еру transmission with two energies Е1 and Е2 yields a system of equations for two unknown concentrations:
(- ln T1) / x = M1 = μ11 × ρ1 + μ12 × ρ2 |
|
(5.12) |
|
. |
|
(- ln T2 ) / x = M 2 = μ12 × ρ1 + μ22 × ρ2 |
|
|
The solution to the equations looks as follows:
246
ρ1 |
= (M1 × μ22 - M 2 |
× μ21 ) / D |
(5.13) |
|
|
, |
|
ρ2 = (M 2 × μ11 - M1 × μ12 ) / D |
|
||
where D = μ11 × μ22 - μ12 × μ12 .
The more accurate is the solution, the more nonzero is the quantity D. This condition is fulfilled if two radiations near and on both sides of the heavy mixture component absorption K-edge (or L-edge) are used. With even the absorption factors of a light components differing slightly at these energies, the difference of D will be rather great.
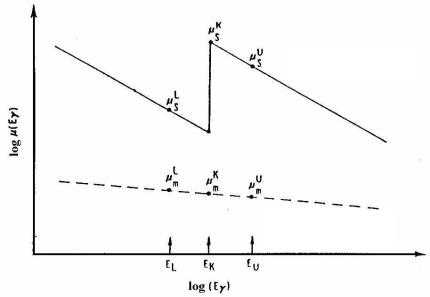
We shall consider in detail the changes of μ (Е) near the К-electron absorption edge (Fig. 5.10).
Heavy component material
Light component material
Fig. 5.10. Changes in the absorption factors of a heavy and a light NM component near the K-edge
By measuring the transmission of γ-radiations with the energies of ЕL and ЕU, we find the concentration of NM in the solution ρs sought for:
247
ρs = |
1 |
ln( |
TL |
) + ρm ( Dμm ) , |
(5.14) |
|
DμS × x |
TU |
|||||
|
|
Dμs |
|
where Dμs = μsU - μsL >0, Dμm = μmU - μmL <0.
If ЕL and ЕU are very close to ЕK, then Dμm is tending towards zero, and the measurement result is insensitive to the absorption in the matrix.
Extra measurements (calibrations) are done with reference solutions placed in standard containers.
If NM concentrations are small (dozens of g/l), L-edge densitometry is used; where higher NM concentrations are involved (hundreds of g/l), K- edge densitometry is employed.
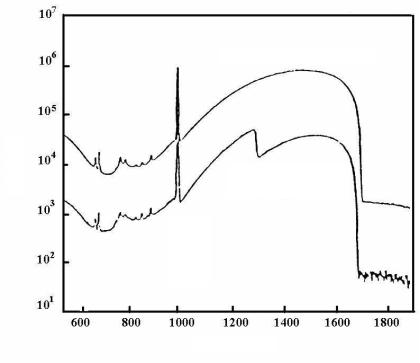
Two types of radiation sources are used: the first one is X-ray generators that produce photons with a continuous broad spectrum. These are used both in K-edge and L-edge measurements. Examples of spectra are given in Fig. 5.11. The relation of the sample spectrum to the comparison spectrum gives the transmission.
The second type of employed sources is represented by radioactive g- sources of monoenergy radiations. For example, plutonium solutions (Ек=121.82 keV) are measured using two sources: 75Se (Т1/2=120 days,
Еγ=121.1 keV) and 57Со (Т1/2=270 days, Еγ=122 keV). The result of the absorption in a light-weight matrix during measurements with these sources is influenced negligibly, still different decay rates thereof should be taken into account.
Measurements of uranium solutions (Ек = 115.61 keV) use the ytterbium-196 (169Yb) g-source with a half-life of Т1/2 = 32 days. In this case, no allowance for decay is required to be made because one source emits both quanta groups. However, a major difference in the energies thereof (EL = 109.8 keV, EU = 130.5 keV) causes a great sensitivity to the absorption in the matrix (high Dμm).
248
Count
Comparison spectrum
Uranium spectrum 197 g/l
88.04 keV Cd-109
Channels
Fig. 5.11. Spectrum of solutions obtained with the aid of an X-ray generator: the upper curve is the comparison spectrum measured by a K-edge densitometry detector in an NM-free solution (3-mol solution of HNO3); the lower curve is the spectrum of an uranium solution sample with a concentration of 197 g/l; the 109Cd source is used to correct the counting losses caused by dead-time
L-edge analyses are used for samples of a smaller thickness, this being dictated by a smaller penetrating power of the radiations measured. L-edge densitometry (LED) is normally used to control solutions with smaller concentrations of NM, and K-edge densitometry (KED) is used for higher concentrations (Table 5.9). With lower NM concentrations in solutions, an X-ray fluorescence analysis is used.
Table 5.9
Representative NM concentrations in solutions for densitometry analyses
Representative concentrations, g/l |
Uranium |
Plutonium |
ρS (KED) |
270 |
294 |
ρS (LED) |
18 |
19 |
249 |
|
|
Analyses of NM solutions. X-ray fluorescence analysis (XFA)
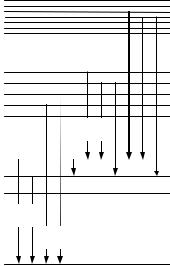
All atoms have ordered systems of electron shells that are characterized by particular values of binding energy. Where a vacancy emerges on an electron shell, it is filled with another electron passing from a higher shell (Fig. 5.12). The difference of potential energies is liberated as an X-ray quantum with the energy equal to the difference of binding energies on the lower and the upper shells. The characteristic radiation spectra are fairly simple, this making it easier to analyze these.
To release the electron from the shell, one needs to give it the energy in excess of Еbinding. To do this, outer excitation sources are used.
The probability of exciting an X-ray source of a K (or L) series depends on the difference between the excitation source quanta energy Е0 and the K–(or L)–shell electron binding energy Еbinding. The excitation probability reaches the maximum when the gamma-quantum energy exceeds the electron binding energy just slightly.
VII
N
I
|
L α1 |
α2 |
|
|
|
|
|
|
|
|
|
γ1 |
|
||
|
β1 |
β2 |
|
|
|||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
V
K α1 α2
β3 β1
M
Fig. 5.12. Diagram of the electron transitions in an atom
Radioactive sources or X-ray tubes are used to excite characteristic radiation. The advantages of radioactive sources are a simple design and stability of the exciting radiation energy. The exciting radiation spectrum
250