

Хенке_Жидкостная хроматография [2009]
.pdf8.3. Препаративная гелевая хроматография 211
киванию «разрезанных» пиков смесь можно разделить количественно вплоть до геп! тасахарида. Количественную оценку и соответственно препаративное выделение отдельных компонентов можно было бы провести на готовых стеклянных колон! ках Lobar®!LiChroprep®NH2, фирмы Мерк/Дармштадт следующим образом:
•с помощью чистого ацетонитрила на 6 последовательно соединенных ко! лонках, количество пробы/цикл разделения: 3…5 г,
•смесью ацетонитрила и воды 65 : 35 на 1…2 колонках. Величина пробы мо!
жет достигать 1…2 г.
Были выбраны следующие условия разделения (см. рис. 8.25).
ГЛАВА 9
ИНСТРУКЦИЯ ПО ПРОВЕДЕНИЮ АНАЛИЗОВ
9.1. Переэтерификация
9.1.1. Принцип
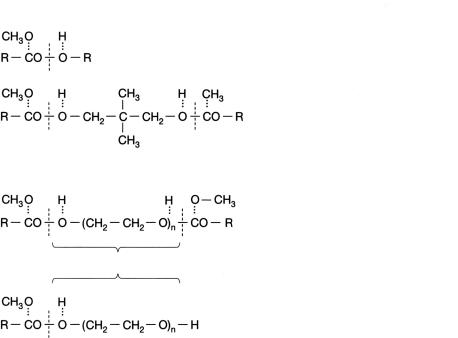
Компоненты спирта выделяются в свободном виде из различных сложных эфиров жирных кислот одно! и многоатомных спиртов, сложных эфиров дикарбоновой кис! лоты, сложных эфиров полигликоля жирной кислоты, а также из алифатических и ароматических сложных полиэфиров благодаря переэтерификации одноатомными
спиртами с короткой цепью (алкоголиз). Жирные кислоты, алифатические, а также ароматические дикарбоновые кислоты представлены как сложные эфиры дикарбо! новой кислоты или сложные моноэфиры одноатомных спиртов. При алкоголизе раз! личных сложных полиэфиров и сложных эфиров полигликоля жирной кислоты мож! но говорить о деполимеризирующей переэтерификации. Рисунок 9.1 показывает пе! реэтерификацию сложных эфиров жирных кислот одно! и двухатомных спиртов, а
1. Переэтерификация сложных эфиров кислоты жирного ряда одно и многоатомных спиртов, например:
1 моль сложного метилового эфира кислоты + 1 моль спирта
2 моля сложного метилового эфира жирной кислоты + 1 моль спирта (диол)
2. Переэтерификация аддуктов ЭО кислоты жирного ряда
2 моля сложного метилового эфира
жирной кислоты + 1моль ПЭГ
(сложный диэфир)
ПЭГ
1 моль сложного метилового эфира
жирной кислоты + 1 моль ПЭГ
(сложный моноэфир)
Рис. 9.1. Переэтерификация различных сложных эфиров жирного ряда с помощью метанола

9.1. Переэтерификация 213
также аддуктов ЭО жирной кислоты (сложные эфиры полигликоля жирной кисло! ты), которые применяются как компоненты прядильной препарации. Само собой разумеется, что переэтерификация и деполимеризирующий алкоголиз сложных эфи! ров дикарбоновой кислоты или сложных полиэфиров ароматического или алифати!
ческого происхождения осуществляются по одной и той же реакционной схеме.
9.1.2. Проведение
Переэтерификация или алкоголиз может осуществляться с помощью метанола, этанола, 2!пропанола (изопропанол) или 1!бутанола. Соответственно имеются моно! и дикарбоновая кислоты в качестве сложного метилового, этилового, изо! пропилового или 1!бутилового эфира.
1 г пробы с 20 мг спирта + 10 мг ацетата Zn (катализатор переэтерификации) проходят переэтерификацию в течение 24 часов при 230°С в автоклавах из специ!
альной стали под высоким давлением при наличии 50 мл тефлонового наполни! теля (фирма Бергхоф/Тюбинген). Если в качестве подвижной фазы использовал! ся метанол (а это, как правило, так и бывает), можно после охлаждения автоклава
напрямую, т. е. не удаляя средство для переэтерефикации, подавать метанолизат на разделение. Высшие спирты перед разделением удаляют на ротационном ис!
парителе или еще лучше путем дистилляции.
9.1.2.1. Аналитическое разделение и количественная оценка
Смеси сложных метиловых эфиров кислоты жирного ряда, сложные диметило! вые эфиры дикарбоновой кислоты, а также различные одно! и многоатомные
спирты, как и выделенные в несвязанном виде полиэтиленгликоли, можно раз! делять с помощью аналитической ВЭЖХ или ГХ, определять количественно и
одновременно идентифицировать на основе времени удерживания. Аналитичес! кая ВЭЖХ, как правило, не может отделять смеси веществ разной полярности одной системой разделения (неподвижная + подвижная фаза). Если используют
ОФ!материалы, то сложные метиловые эфиры кислоты жирного ряда, сложные диметиловые эфиры дикарбоновой кислоты разделяются при меньшим количе! ством воды в метаноле, чем свободные спирты. Для различных ПЭГ, таких как
200, 400, 600 или выше, требуется еще больше воды в метаноле, для того чтобы
хорошо разделить смесь олигомеров и тем самым можно было идентифицировать на основе пика стандарта.
9.1.2.2. Препаративное разделение и количественная оценка
Препаративное разделение, выделение и одновременная количественная оценка производится на ОФ!материалах на готовых стеклянных колонках низкого дав!
ления с помощью чистого метанола, т. е. метанолизат можно сразу, как только
извлекли из автоклава, подавать на колонку. Препаративное разделение имеет су!
щественные преимущества по сравнению с аналитической ВЭЖХ. Сложные ме!
тиловые эфиры кислоты жирного ряда, сложные диметиловые эфиры дикарбо! новой кислоты, а также высшие спирты, такие как додеканол, тетрадеканол или октадеканол, можно сразу оценивать количественно по результатам взвешивания.
Полиэтиленгликоль, выделенный в свободном виде из аддуктов ЭО жирной кисло!
214
 Глава 9. Инструкция по проведению анализов
Глава 9. Инструкция по проведению анализов
ты, также можно определить точно с помощью гравиметрического способа. Иден! тификация осуществляется посредством ВЭЖХ на колонках ОФ смесью метано! ла и воды. Катализатор переэтерификации (ацетат цинка) распределяется в мета!
нолизате в виде коллоида и появляется при разделении на метериалах ОФ перед
несвязанным ПЭГ. Ввиду того, что ацетат цинка обладает ионогенными свойства! ми, он исключается, т. е. он проникает в систему пор лишь незначительно.
9.2.Дериватизация через этерификацию
Вгазовой хроматографии дериватизация проводится уже в течение многих лет для того, чтобы перевести в стабильные дериваты с трудом испаряющиеся или разлагающиеся соединения. Именно свободные органические кислоты, будь то жирные, дикарбоновые кислоты или гидроксикислоты, являются сильно поляр!
ными и поэтому трудно испаряющимися. С самого начала газовой хроматогра! фии кислоты преобразовывали спиртами и таким образом получали легколету!
чие сложные эфиры. Более всего распространено изготовление сложного мети! лового эфира с помощью следующих методов этерификации:
А: кислота + спирт + BF3 в качестве катализатора В: кислота + спирт + HCl в качестве катализатора
C:кислота + спирт + H2SO4 в качестве катализатора
D:кислота + диазометан
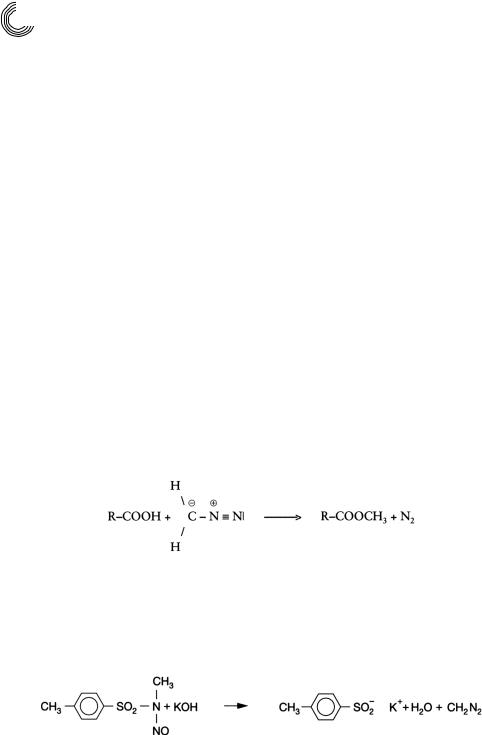
МЕТОД ЭТЕРИФИКАЦИИ D
При этом способе этерификации органическая кислота с помощью диазометана пре! вращается в сложный метиловый эфир [9.1] по следующему уравнению реакции.
9.2.1. Получение раствора простого эфира диазометана
N!нитрозо!толуол!4!сульфометиламид с помощью калийной щелочи, растворен!
ной в этаноле, преобразовывается в соль калия толуол!4!сульфоновой кислоты, и при этом диазометан выделяется в несвязанном виде по следующему уравнению
реакции, который передистиллируется простым диэтиловым эфиром.
Этим раствором преобразовывают органическую кислоту или смесь кислот. Количественное превращение устанавливается следующим образом: избыточный
диазометан распознают по свойственной желтой окраске и потому, что газовые
пузырьки (N2) больше не поднимаются.
9.3. Катионит 215
9.3. Катионит
Катиониты используют в том случае, если в несвязанном виде выделяют кислот!
ные соединения из солей щелочных металлов. Это могут быть соли щелочных металлов различных карбоновых или фосфорных кислот, которые после удале!
ния щелочных ионов легко могут превратиться в производные. Этот процесс имеет и другие преимущества; производные кислот, например различные сложные эфи! ры, лучше растворимы в органических растворителях. Лучшая растворимость в
легколетучих органических растворителях обозначает, что в препаративном мас!
штабе регенерация подвижных фаз происходит более простым, быстрым и более
щадящим способом, чем удаление воды в довольно больших количествах.
9.3.1. Проведение
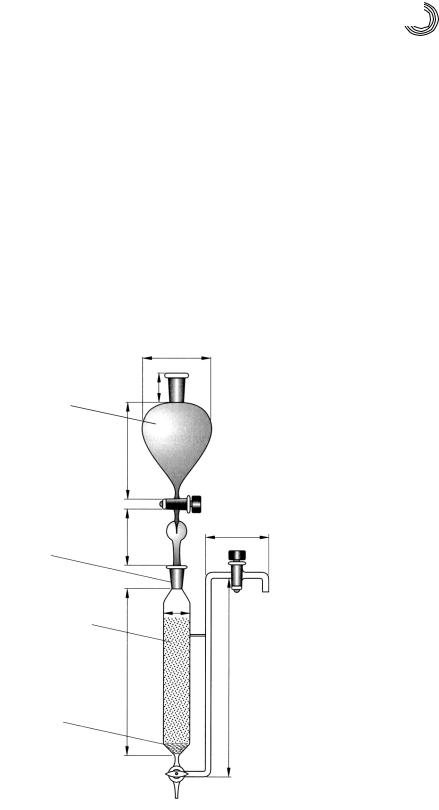
Катионит, в данном случае сильнокислый ионообменник I фирмы Мерк (Арт.!№ 4765),
регенерируется и приводится в исходное состояние с помощью 100 мл 2n HCl и
затем промывается дистиллированной водой до нейтрального состояния. Перед
70 мм |
Рис. 9.2. Колонка ионо! |
|
обменника |
30 мм
Капельная воронка
100 мм
мм |
70 мм |
60 |
|
NS 14,5 – стандартный |
|
(нормальный) шлиф |
|
|
30 мм |
Ионообменник (ионит) |
|
180 мм |
200 мм |
Кварцевый войлок
NS: нормальный (стандартный) шлиф Колонка ионообменника

216
 Глава 9. Инструкция по проведению анализов
Глава 9. Инструкция по проведению анализов
проведением ионообменного процесса он промывается смесью, состоящей из 48 ча! стей дистиллированной воды и 12 частей этанола. На рис. 9.2 изображена колонка ионообменника с капельной воронкой.
500…1000 мг пробы растворяют в смеси (примерно 30 мл) дистиллированной
воды и этанола и смывают в капельную воронку. Сливной кран открыт лишь час! тично, чтобы раствор пробы стекал медленно на катионит. Одновременно краны на колонке соответственно открыты для улавливания элюата в колбе Эрленмейе!
ра (коническая колба). Затем следует повторная промывка смесью дистиллиро! ванной воды и этанола (примерно 50 мл). Собранный элюат лучше всего просу!
шить при 100°С в камерной сушилке с циркуляцией воздуха.
Несвязанные карбоновую или фосфорную кислоты можно преобразовать в соответствующие сложные эфиры посредством раствора диазометана в эфи! ре или другим методом этерификации. Количественный ход метилирования с помощью раствора диазометана в эфире можно определить следующим об! разом:
•нет образования пузырьков (N2, см. раздел 9.2, метод этерификации D),
•сохраняется желтый цвет раствора.
9.4. Определение общего количества этиленоксида
Описываемый ниже метод удобен для определения общего количества звеньев (–СН2–СН2–О–) в аддуктах этиленоксида.
9.4.1. Принцип химико аналитического метода
Йодистоводородная кислота получается как промежуточным продукт из йодида
калия и фосфорной кислоты:
H3PO4 + 3 KJ → 3 HJ + K3PO4.
Эта йодистоводородная кислота сразу же образует этилендийодид с группами
(–СН2СН2О):
(–СН2 –СН2–О–)х + 2 · HJ = x · (JCH2–CH2J) + x · H2O.
Из!за избытка HJ образуется этилйодид и йод:
J–CH2–CH2–J + HJ = CH3–CH2–J + J2.
Этиленйодид распадается на этилен и йод при температуре 120°С:
J–CH2–CH2–J → CH2 = CH2 + J2.
Освободившееся количество йода эквивалентно группам (–СН2–СН2–О–)х и
титруется 0,1n Na2S2O3.
9.4. Определение общего количества этиленоксида 217
J2 |
|
+ 2 е |
|
|
→2 |
J′ |
|
2 S O 2– |
– 2 е |
|
→ S O |
2– |
|||
2 |
3 |
|
|
|
4 |
6 |
|
J + 2 S O 2– |
→ S O 2– |
+ 2 J′ |
|||||
2 |
2 |
3 |
4 |
6 |
|
|
|
Этот метод описал К. Обруба (K. Obruba) [9.2] в 1961 и 1964 годах, но он нис!
колько не утратил своей актуальности. Часто бывает так, что с помощью 1Н!ЯМР невозможно определить точное количество групп (–СН2–СН2–О–) в смесях. Точ! ное описание определения с аппаратурой и реагентами можно прочитать в [9.2].
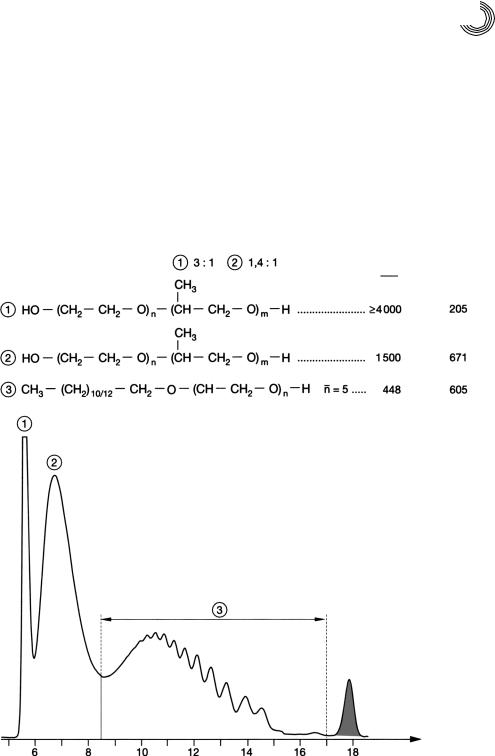
Рисунки 9.3 и 9.4 показывают препаративное гель!хроматографическое раз!
деление аддуктов ЭО/РО и ЭО, а также разделение растительных кислот.
* Молярное соотношние ЭО/ПО
ММ мг
* Согласно 1Н ЯМР спектру
Вода = Vz + Vp
ч
Рис. 9.3. Препаративное гель!хроматографическое разделение аддуктов ЭО/ПО и аддуктов ЭО спирта жирного ряда (Сефадекс LH!20, 5 м, внутренний диа! метр 25/26 мм, метанол 2 мл/мин)
218
 Глава 9. Инструкция по проведению анализов
Глава 9. Инструкция по проведению анализов
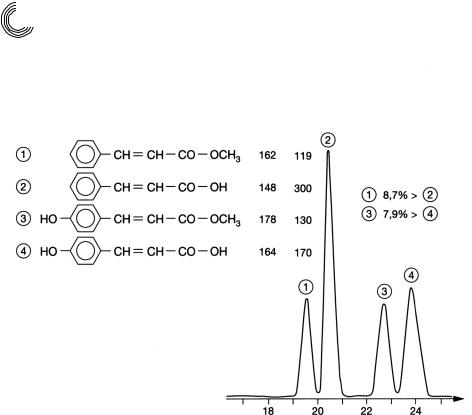
–Коричная кислота и сложный метиловый эфир коричной кислоты  соответственно
соответственно 
–р кумаровая кислота и сложный метиловый эфир р кумаровой кислоты  соответстветственно
соответстветственно 
Ароматическая группа – ОН сильно адсорбирована!!
ММ мг
ч
Рис. 9.4. Препаративное гель!хроматографическое разделение растительных кис! лот (природные вещества): условия разделения, как на рис. 9.3

Сокращения и символы
Сокращения не следуют строгим правилам, когда применяются немецко! и анг! лоязычные обозначения. В этой форме их находят также в монографиях и публи! кациях.
СССцентробежная противоточная хроматография (ЦПХ)
DC |
тонкослойная хроматография (ТХ) |
DCCC |
противоточная капельная хроматография (ПКХ) |
GC |
газовая хроматография (ГХ) |
CLC |
газожидкостная хроматография (ГЖХ) |
GPC |
гель!проникающая хроматография (ГПХ) |
GSC |
газоадсорбционная хроматография (ГАХ) |
HPLC |
высокоэффективная жидкостная хроматография (ВЭЖХ) |
HPTLC |
высокоэффективная тонкослойная хроматография (ВЭТХ) |
IR |
инфракрасная спектроскопия (ИК) |
LC |
жидкостная хроматография (ЖХ) |
MS |
масс!спектрометрия (МС) |
NMR |
ядерная магнитно!резонансная спектроскопия (ЯМР) |
PC |
бумажная хроматография, хроматография на бумаге (БХ) |
PEG |
полиэтиленгликоль (ПЭГ) |
PUR |
полиуретан (ПУР) |
RLCC |
распределительная хроматография вращения (ротационная) (РХ) |
SC |
колоночная хроматография (КХ) |
SFC |
хроматография со сверхкритическими подвижными фазами (СХ) |
5 м (1 м + 2 по 2 м, внутренний диаметр 25/26 мм) с помощью метанола,
2 мл/мин
5 м (1 м + 2 по 2 м, внутренний диаметр 25/26 мм) с помощью ацетона,
4 мл/мин
Звезда и заполненный треугольник обозначают, что разделения проводятся на идентичных колоночных наборах при полностью одинаковых условиях разделения.
Длина слоя геля при 5!метровых колонках с внутренним диаметром 25/26 мм со! ставляет: 870 + 2 × 1870 мм = 461 см.
Литература
[4.1] Henke, H.: LaborPraxis, Februar 1995, S. 62.
[4.2] Schönfeldt, N.: Grenzflächenaktive Ethylenoxid'Addukte. WVG Stuttgart, 1976. [4.3] Schönfeldt, N.: Ergänzungsband, a.a.O., 1984.
[4.4] Henke, H.: LaborPraxis, Februar 1995, S. 62.
[4.5] Henke, H.: Präparative Gelchromatographie an Sephadex LH'20. Heidelberg: Hüthig Verlag, 1994.
[4.6] Henke, H.: LaborPraxis, Februar 1995, S. 62.
[4.7] Stahl, E. (Hrsg.): Chromatographische und mikroskopische Analyse von Drogen. Stuttgart: Gustav Fischer Verlag, 1970.
[4.8] Henke, H.: Präparative Gelchromatographie an Sephadex LH'20. Heidelberg: Hüthig Verlag, 1994.
[4.9] Unger, K. K. (Hrsg.): Handbuch der HPLC, Teil 1. Darmstadt: GIT Verlag, 1989.
[4.10] Henke, H.: Präparative Gelchromatographie an Sephadex LH'20. Heidelberg: Hüthig Verlag, 1994.
[4.11] Winkle, W.: Chromatographia, Vol. 29, S. 530, 1990.
[5.1] Hein, H.: Spinnpräparationen für synthetische Fäden, Deutscher!Färber!Kalender, 1974, S. 295–301.
[5.2] Egginger, R.: Analysenschema zur Identifizieruilg der chemischen Gruppenzugehörigkeit von Textilhilfsmitteln (Tensidgruppenstoffen). Textilbetrieb, August 1979, S. 49–51.
[5.3] Mutschler, G.: Spinnpräparationen für die Friktionstexturierung. Chemiefasern Textil' industrie, Februar 1981, S. 131–134.
[5.4] Schollmeyer, E., Deuter, U.: Entwicklung eines Trennungsganges für die einfache Analyse typischer Inhaltsstoffe von Textilhilfsmitteln in Flotten und auf Textilien. Deutsches Textil! forschungszentrum, Krefeld, Dezember 1991.
[5.5] Schönfeldt, N.: Grenzenflächenaktive Ethylenoxid!Addukte, WVG Stuttgart, 1976. [5.6] Schönfeldt, N.: Ergänzungsband, a.a.O., 1984.
[5.7] Henke, H.: HPLC!Trennung von Fettsaureestern 1! und mehrwertiger Alkohole. Vortrag beim HPLC!Seminar der Fa. Knauer/Berlin, 1979.
[5.8] Henke, H., Schubert, J.: HRC & CC, Vol. 3, Februar 1980, S. 69–77. [5.9] Henke, H., Dietrich, W.: HRC & CC, Vol. 3, Juni 1980, S. 277–285. [5.10] Henke, H., Rülke, K.: SWISS CHEM 9 (1987) Nr. 7/8, S. 23–30.
[5.11] Henke, H.: 1. Arbeitstagung präparative HPLC, Monheim (D), 19./20. Januar 1987. [5.12] Henke, H.: Tenside Detergents 15 (1978) 193.
[5.13] Henke, H.: LaborPraxis, 4 (1980) 62.
[5.14] Henke, H.: LaborPraxis, 2 (1995) 62.
[6.1] Bundesgesundheitsblatt 23, Nr. 22 v. 31.10.1980, 45. [6.2] Mitteilung. Frauck: Kunststoffe 28, Lfg., März, 1981.
[7.1] Corish, P. J., Davison, W. T. H.: J. Chem. Soc. 1955, S. 2431. [7.2] Mulder, I. L.: Anal. Chim. Acta, 38, S. 563 (1967).
[7.3] Dawson, B., Hopkins, S., Sewell, P. R.: J. Appl. Polymer Sci. 14,1970, S. 35. [7.4] Wittendorfer, R. E.: Anal. Chem. 36,1964, 960.
[7.5] Winterscheid, H.: Seifen, Öle, Fette Wachse, 80, 1954, S. 404.
[7.6] Majewska, F., Spawozdanie, I. T. S.: Analyza tworzyw poliuretanowych metoda chromato!graphii jonitowej (I. T. S. Report, Analysis of Polyurethanes by Jonite Chromatography), 1962.
[7.7] Schröder, E.: Plastic und Kautschuk 9,1962, S. 186. [7.8] Fanica, L.: Chim. Anal. (Paris) 38, 1956, S. 360.