

Требования к неопределенности измерений
Испытания |
Требования |
1 |
2 |
Количественное определение: Субстанции
Другие субстанции и готовые лекарственные препараты |
|
Однородность содержания, растворение |
|

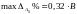

Продолжение табл. 2.3
1 |
2 |
Сопутствующие примеси |
Предельные
тесты
Количественные
тесты
|
Остаточные органические растворители |
|


Для расчета полной неопределенности (ΔAs), могут быть использованы различные подходы, среди которых можно выделить два основных: линейная модель и подход Уэлча-Сатэртуэйта.
Линейная
статистическая модель.
Если измеряемая величина у является
функцией n
независимых случайных величин xi,
то дисперсия величины у связана с
дисперсиями величин
 соотношением:
соотношением:
 ,
(2.60)
,
(2.60)
где
 – частная производная функции по
аргументу xi.
Является весовым коэффициентом,
показывающим как величина у
изменяется с изменением значения
величины xi.
– частная производная функции по
аргументу xi.
Является весовым коэффициентом,
показывающим как величина у
изменяется с изменением значения
величины xi.
Обобщением соотношения (2.61) является следующее соотношение:
 ,
(2.61)
,
(2.61)
т.е.
доверительный интервал функции Δy
связан с доверительными интервалами
переменных
 выражением 2.61.
выражением 2.61.
В фармакопейном анализе y представляет собой обычно произведение или частное случайных и постоянных величин (масс навесок, разбавлений, оптических плотностей или площадей пиков и т.д.):
 ,
(2.62)
,
(2.62)
В этом случае весовые коэффициенты принимаются равными единице и получаем:
 .
(2.63)
.
(2.63)
В ЕС и США для расчета неопределенности функции нескольких случайных переменных широко применяется статистическая модель Уэлча-Сатуртуэйта. Данный подход дает несколько более узкие доверительные интервалы, чем линейная модель. В то же время, если методика валидирована с использованием линейной статистической модели, то она тем более будет соответствовать валидационным критериям модели Уэлча-Сатуртуэйта, поскольку ее критерии будут более либеральны, чем полученные для линейной модели.
При проведении
фармакопейного анализа в суммарной
неопределенности (
)
анализа обычно можно выделить такие
типы неопределенности: неопределенность
пробоподготовки ( ),
неопределенность конечной аналитической
операции (
),
неопределенность конечной аналитической
операции ( )
и неопределенность аттестации стандартного
образца (
)
и неопределенность аттестации стандартного
образца ( ).
Величина неопределенности стандартного
образца обычно мала и ею можно пренебречь.
).
Величина неопределенности стандартного
образца обычно мала и ею можно пренебречь.
Учитывая это, прогнозируемая полная неопределенность результатов анализа не должна превышать максимально допустимого значения max . Она рассчитывается по формуле:
 .
(2.64)
.
(2.64)
где – неопределенность пробоподготовки (прогноз); – неопределенность конечной аналитической операции (прогноз).
Неопределенность пробоподготовки может быть рассчитана по формуле линейной статистической модели:
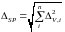 .
(2.65)
.
(2.65)
где
 –
составляющая неопределенности, связанная
с конкретной операцией пробоподготовки
(взятие навески, аликвоты малого объема,
доведение до объема в мерной колбе и
др.), выраженная как односторонний
относительный доверительный интервал
для уровня доверия 0,95.
–
составляющая неопределенности, связанная
с конкретной операцией пробоподготовки
(взятие навески, аликвоты малого объема,
доведение до объема в мерной колбе и
др.), выраженная как односторонний
относительный доверительный интервал
для уровня доверия 0,95.
Прогнозируемая неопределенность конечной аналитической операции ( ) для хроматографических методик может быть рассчитана из требований к относительному стандартному отклонению в испытании на пригодность хроматографической системы (RSDmax) и используемого числа хроматограмм в методике.
Для спектрофотометрических методик при прогнозе неопределенности конечной аналитической операции рекомендуется исходить из относительного стандартного отклонения оптической плотности с рандомизацией положения кювет, полученного в межлабораторном эксперименте (0,52%) , и числа параллельных измерений (рекомендуется при выполнении анализа и при оценке неопределенности принимать, что число измерений должно быть не менее 3).
Учитывая наличие 2 растворов (испытуемого и раствора сравнения), и рекомендации проводить не менее 3 параллельных измерениях оптической плотности с выниманием кюветы для каждого раствора, для спектрофотометрического анализа расчет производится в соответствии с формулой 2.66:
 ,
(2.66)
,
(2.66)
где 1,65 – коэффициент Гаусса для односторонней вероятности 95%.
Выражение (2.65) характеризует ту неопределенность конечной аналитической операции спектрофотометрического анализа, которая характерна в настоящее время для отечественной системы государственных лабораторий контроля качества ЛС.
Рекомендуется, чтобы при разработке методик прогнозируемая неопределенность пробоподготовки была незначима, т. е. выполнялось соотношение:
 .
(2.67)
.
(2.67)
Данное соотношение не всегда может быть выполнимо. Так, в случае обычной спектрофотометрии основным источником неопределенности результатов является, как правило, пробоподготовка. Однако для хроматографических методик предполагается, что обычно основным источником неопределенности является конечная аналитическая операция. Если неопределенность (прогнозируемая или изученная экспериментально), связанная с пробоподготовкой, является значимой, то к неопределенности конечной аналитической операции следует предъявлять, соответственно, более жесткие требования, чтобы обеспечить выполнение критериев для полной неопределенности результата анализа.
Робастность (устойчивость).
Робастность является показателем надежности МИ при изменении условий работы или окружающей среды (которые, тем не менее, совместимы с нормальными условиями).
Оценка робастности проводится при разработке МИ.
Влияние условий проведения испытания на результаты анализа должно контролироваться, а текст МИ должен содержать соответствующие предупреждения.
На результаты испытаний оказывают влияние следующие основные факторы:
стабильность растворов, используемых в аналитических методах;
время экстракции;
варьирование времени инкубации;
незначительное отклонение рН реактива;
колебания температуры или влажности внутри помещения или термостата.
Для жидкостной хроматографии примерами влияющих факторов являются:
изменение рН подвижной фазы;
изменение состава подвижной фазы;
различные колонки (разные серии и поставщики);
температура;
скорость подвижной фазы.
Для газовой хроматографии примерами влияющих факторов являются:
различные колонки (разные серии и поставщики);
температура;
скорость газа-носителя.
Одним из следствий оценки робастности является определение серии параметров пригодности системы (например, испытание на разделение), которые обеспечивают корректность МИ во всех случаях ее использования. Оценка робастности предопределяет установление ряда параметров пригодности системы.
Ориентировочный перечень вопросов, на которые должны быть даны исчерпывающие ответы при определении робастности:
является ли данный метод измерений эффективным при разных условиях проведения (реактивы из разных источников, различные операторы, различные дни анализа);
изменения каких параметров метода испытаний оказывают влияние на результаты анализа;
удалось ли доказать воспроизводимость МИ на примере испытаний образцов из нескольких серий.
Стабильность растворов [22].
Изучение стабильности анализируемых растворов является одной из характеристик робастности. Обычно надо показать, что растворы являются стабильными не менее 1 часа. Это означает, что вносимая их нестабильностью систематическая погрешность δ не превосходит предельного значения maxδ.
Критерии стабильности различаются для спектрофотометрии и хроматографии.
В случае спектрофотометрии методом стандарта (испытуемый и стандартный растворы готовят одновременно) надо показать, что изменение отношения оптических плотностей испытуемого и стандартного растворов (в нормализованных координатах это изменение величины Y) не превосходит в течение 1 часа величины maxδ (формулы 2.39−2.40). Для этого параллельно измеряют их оптические плотностей через 0, 15, 30, 45 и 60 минут, определяют величины Y и рассчитывают доверительный интервал ∆Y % . Он не должен превосходить maxδ.
В
случае хроматографического количественного
определения методом стандарта такой
подход принципиально невозможен из-за
длительности хроматографирования (одна
хроматограмма занимает обычно около
20 мин). Однако это имеет и свои преимущества
для доказательства необходимой
стабильности растворов. Действительно,
если приготовили и провели хроматографирование
10 растворов для изучения линейности,
то это время значительно превышает
время анализа 1−3 образцов, которые
обычно анализируют на практике. Поэтому
положительные результаты точности и
правильности, полученные при изучении
линейности, являются подтверждением
необходимой стабильности растворов.
Другим доказательством является
практически незначимое различие ( )
величин Z
для первого и последнего хроматографируемого
растворов.
)
величин Z
для первого и последнего хроматографируемого
растворов.
Лабораторная работа № 15
 ОПРЕДЕЛЕНИЕ
ВАЛИДАЦИОННЫХ ХАРАКТЕРИСТИК
ОПРЕДЕЛЕНИЕ
ВАЛИДАЦИОННЫХ ХАРАКТЕРИСТИК
И ПОКАЗАТЕЛЕЙ ТОЧНОСТИ КОЛИЧЕСТВЕННОГО
ОПРЕДЕЛЕНИЯ НАТРИЯ УКСУСНОКИСЛОГО 3-ВОДНОГО В ЛЕКАРСТВЕННОМ ПРЕПАРАТЕ «АЦЕСОЛЬ, РАСТВОР ДЛЯ ИНЪЕКЦИЙ»
Цель работы: провести валидацию методики количественного определения натрия уксуснокислого 3-водного в лекарственном препарате «Ацесоль, раствор для инфузий».
Средства испытаний: весы лабораторные AS220/C/2 с точностью взвешивания 0,1 мг; кислота хлористоводородная, раствор с концентрацией 0,1 М; раствор бромфенолового синего; натрия ацетат тригидрат (натрий уксуснокислый 3-водный) (НД РБ 0559С-2011, ООО НПФ «Невский химик», РФ; натрий хлорид (НД РБ 0051С-2010, ТД ООО «Малиновое озеро», РФ); калия хлорид (НД РБ 0598С-2010, ОАО «Востоквит», РФ); вода для инъекций; лабораторная посуда.