

Примеры валидационных критериев для разных допусков
содержания числа точек n = 9
Испытание |
Диапазон D, шаг, RSDy, % |
B,% |
max ΔAs, % |
max δ, % |
max RSD0, % |
min r |
max a, % |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
Субстанции |
|||||||
КО |
D=80−120, Шаг =5 RSDy = 13,69 |
1,0 |
1,0 |
0,32 |
0,53 |
0,99926 |
1,6 |
1,5 |
1,5 |
0,48 |
0,79 |
0,99833 |
2,4 |
||
2,0 |
2,0 |
0,64 |
1,06 |
0,99702 |
3,2 |
||
2,5 |
2,5 |
0,80 |
1,32 |
0,99535 |
4,0 |
||
3,0 |
3,0 |
0,96 |
1,58 |
0,99329 |
4,8 |
||
Готовые лекарственные средства |
|||||||
КО |
D=80−120, Шаг =5 RSDy = 13,69 |
5 |
1,60 |
0,51 |
0,84 |
0,99810 |
2,6 |
7,5 |
2,4 |
0,77 |
1,27 |
0,99571 |
3,8 |
||
10 |
3,2 |
1,02 |
1,69 |
0,99236 |
5,1 |
||
15 |
4,8 |
1,54 |
2,5 |
0,98273 |
7,7 |
||
20 |
6,4 |
2,1 |
3,4 |
0,96909 |
10,2 |
||
ОС |
D=70−130, Шаг =7,5 RSDy = 20,54 |
_ |
3,0 |
0,96 |
1,58 |
0,99710 |
3,1 |
Р |
D=55−135, Шаг =10 RSDy = 27,39 |
_ |
3,0 |
0,96 |
1,58 |
0,99710 |
3,1 |
Правильность.
Правильность должна быть подтверждена для всего диапазона применения МИ. Показателем правильности является лабораторное смещение.
Оценка правильности при количественном определении.
Активная субстанция.
Могут быть использованы следующие варианты для оценки правильности при определении содержания или активности фармацевтической субстанции:
− применение МИ к образцам с известной степенью чистоты в соответствии с ТНПА на данную фармацевтическую субстанцию (по возможности такие образцы кроме определяемого вещества должны содержать все компоненты фармацевтической субстанции);
− сравнение результатов анализа, полученных с использованием валидируемой МИ, и результатов, полученных с помощью арбитражной методики, правильность которой определена.
Для биологических субстанций правильность можно определить не всегда по причине отсутствия чистых образцов для анализа. В этом случае результаты сравнивают с характеристиками СО (первичного или вторичного), который параллельно анализируется в рамках одного и того же испытания.
Готовый лекарственный препарат.
Существуют следующие варианты для оценки правильности при определении содержания или активности лекарственных средств:
− применение МИ к искусственным смесям компонентов лекарственного средства, приготовленным в соответствии с ТНПА на данное лекарственное средство и в количествах ±10% от регламентируемого ТНПА содержания;
− при отсутствии образцов всех компонентов лекарственного средства применимо или добавление известных количеств определяемого компонента в испытуемый образец (пробу), или сравнение с результатами, полученными с помощью арбитражной методики, правильность которой определена;
− заключение о правильности должно быть сделано после определения прецизионности, линейности и специфичности.
Примеси (количественное определение).
− правильность МИ по определению количественного содержания примесей определяют на образцах (пробах) (фармацевтической субстанции и лекарственного средства), в которые добавлены известные количества примесей;
− при отсутствии образцов определяемых примесей или продуктов разложения приемлемо сравнение результатов с теми, которые получены с помощью арбитражной методики. Может использоваться фактор отклика лекарственного средства (фармацевтической субстанции).
Должен быть указан конкретный способ нормирования содержания индивидуальной примеси или суммы примесей, например, в массовых процентах, в процентах по отношению к площади пика, но во всех случаях по отношению к основному определяемому компоненту.
Ориентировочный перечень вопросов, на которые должны быть даны исчерпывающие ответы при определении показателя правильности:
− соответствуют ли результаты испытаний пробы, приготовленной из СО, результатам испытаний анализируемой пробы;
− какова степень результативности анализа по выявлению определяемого компонента в любом образце при контроле на примеси;
− какую наименьшую концентрацию примеси, специально вводимой в образец, может обнаружить данный метод испытаний;
− наблюдается ли корреляция между результатами данного метода испытаний и данными арбитражной методики.
Критерии приемлемости.
Правильность можно валидировать одновременно с линейностью и прецизионностью.
В общем случае правильность следует оценивать не менее чем для трех концентраций, входящих в диапазон применения МИ, с использованием как минимум трех полных испытаний для каждой концентрации.
При
обработке серий результатов измерений
необходимо, чтобы результаты входящие
в серию были однородны, т.е. не содержали
выбросов. Проверка однородности выборок
малого объема ( )
осуществляется по Q
тесту [16]. Серию данных упорядочивают
по возрастанию:
)
осуществляется по Q
тесту [16]. Серию данных упорядочивают
по возрастанию:
 .
Для
крайних значений x1
и
xn
(которые
предполагаются выпадающими) рассчитывают
значения контрольного критерия Q
исходя из величины размаха варьирования
R:
.
Для
крайних значений x1
и
xn
(которые
предполагаются выпадающими) рассчитывают
значения контрольного критерия Q
исходя из величины размаха варьирования
R:
для n = 3...7
 ,
(2.29)
,
(2.29)
для n = 8...10
 ,
(2.30)
,
(2.30)
 ,
(2.31)
,
(2.31)
 .
(2.32)
.
(2.32)
Выборка признается неоднородной, если хотя бы одно из вычисленных значений Q1 или Qn превышает табличное значение Q (Р, n), найденное для доверительной вероятности Р. Значения х1 или хn, для которых соответствующее значение Q > Q (P, n) отбрасываются, и для полученной выборки уменьшенного объема выполняют новый цикл вычислений по уравнениям 2.29−2.32 с целью проверки ее однородности.
При
 и
и
 уравнения 2.31 и 2.32 принимают вид
соответственно:
уравнения 2.31 и 2.32 принимают вид
соответственно:
 ,
(2.33)
,
(2.33)
 .
(2.34)
.
(2.34)
Полученная в конечном счете однородная выборка используется для вычисления статистических характеристик.
Правильность можно оценивать по критерию статистической и практической незначимости [17].
Критерий
статистической незначимости.
Систематическую составляющую
неопределенности (смещение,
δ)
можно характеризовать отличием среднего
значения для степени извлечения ( )
от 100%.
)
от 100%.
Среднее значение степени извлечения рассчитывается по формуле:
 ,
(2.35)
,
(2.35)
где
 − измеренное значение содержания
компонента,
– известное значение содержания
компонента.
− измеренное значение содержания
компонента,
– известное значение содержания
компонента.
Смещение статистически неотличимо от нуля, если отклонение от 100% не превышает свой доверительный интервал:
 ,
(2.36)
,
(2.36)
где Δz – односторонний доверительный интервал, рассчитанный по следующей формуле:
 ,
(2.37)
,
(2.37)
где
 − среднее квадратическое отклонение
среднего значения степени извлечения,
рассчитанное по формуле:
− среднее квадратическое отклонение
среднего значения степени извлечения,
рассчитанное по формуле:
 ,
(2.38)
,
(2.38)
где – односторонний коэффициент Стьюдента для доверительной вероятности 0,95 и числа степеней свободы .
Критерий практической незначимости. Если приведенное выше соотношение не выполняется, используют критерий незначимости смещения по сравнению с максимально допустимой неопределенностью анализа:
 .
(2.39)
.
(2.39)
Для субстанции:
 .
(2.40)
.
(2.40)
Для готового лекарственного средства:
 .
(2.41)
.
(2.41)
Из соотношений (2.40−2.41) видно, что критерий практической незначимости зависит только от допусков содержания, но не зависит (в отличие от статистической незначимости) от фактической неопределенности анализа ΔAs. Величины max δ для n = 9 приведены в табл. 2.1.
Для оценки правильность могут быть использованы и другие критерии.
Если известны показатели точности МИ (значения СКО повторяемости и СКО воспроизводимости), то приемлемость смещения может быть оценена по СТБ ИСО 5725-6 (раздел 7, формулы 3, 4) [18].
Для оценки значимости смещения можно использовать критерий Стьюдента [16]:
 ,
(2.42)
,
(2.42)
где
µ – известное значение определяемого
компонента (значение СО, известное
значение добавки);
 –
среднее значение результатов нескольких
измерений, рассчитанное по формуле:
–
среднее значение результатов нескольких
измерений, рассчитанное по формуле:
 ,
(2.43)
,
(2.43)
S – стандартное отклонение результатов измерений:
 .
(2.44)
.
(2.44)
Если
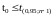 ,
то среднее значение статистически
незначимо отличается от заданного
значения. Если условие не выполняется,
то полученные результаты отягощены
систематической погрешностью δ, %.
Табличное значение коэффициента
Стьюдента находят при доверительной
вероятности 0,95 (двухсторонний доверительный
интервал) и степени свободы
.
,
то среднее значение статистически
незначимо отличается от заданного
значения. Если условие не выполняется,
то полученные результаты отягощены
систематической погрешностью δ, %.
Табличное значение коэффициента
Стьюдента находят при доверительной
вероятности 0,95 (двухсторонний доверительный
интервал) и степени свободы
.
В случае использования арбитражной методики при валидации правильности приемлемость результатов может быть определена с учетом статистических методов проверки гипотезы, например с использованием t и F критериев (формулы 2.51 и 2.55) [16].
Прецизионность
Повторяемость (сходимость).
Повторяемость должна показать, что методика анализа при проведении в одинаковых условиях (как например, один и тот же прибор, тот же лаборант, тот же день, идентичные реактивы) обеспечивает получение сравнимых результатов.
Повторяемость МИ определяется оценкой результатов испытаний идентичных образцов (проб), отобранных из одной и той же серии в условиях повторяемости. Повторяемость определяют, выполняя:
− не менее девяти определений концентраций, входящих в указанный диапазон применения МИ (три концентрации с соответствующим трехкратным повтором всей аналитической методики).
− не менее шести определений для образцов с содержанием анализируемого вещества, близким к номинальному.
Оценка и расчет результатов проводится путем вычисления среднего значения, стандартного отклонения, относительного стандартного отклонения (коэффициента вариации) и доверительного интервала. Критерий приемлемости для коэффициента вариации необходимо установить в зависимости от предполагаемых возможных значений (например, границ нормы для содержания в спецификации ±В). В большинстве случаев коэффициент вариации не должен превышать 1−2 %.
Для проверки приемлемости результатов текущих и контрольных испытаний используют значения предела повторяемости установленных в процессе валидации МИ согласно требованиям СТБ ИСО 5725-6 (раздел 5) [19].
Промежуточная прецизионность (внутрилабораторная воспроизводимость).
С помощью промежуточной прецизионности должно быть доказано, что аналитическая методика обеспечивает получение согласующихся результатов независимо от изменений внешних обстоятельств при условии однородности исследуемой пробы.
Промежуточная прецизионность характеризует влияние вариаций внутри лаборатории. Типичными исследуемыми (вариабельными) факторами согласно СТБ ИСО 5725-3 являются время (Т), оператор (О), оборудование (Е) и калибровка (С) [20]. Нецелесообразно изучать влияние этих факторов в отдельности. Предпочтительно использовать планирование эксперимента в зависимости от условий применимости МИ.
Объем испытаний промежуточной прецизионности зависит от условий, в которых будет применяться данная МИ. Разработчик должен установить влияние случайных факторов на достоверность результатов испытаний.
Для оценки промежуточной прецизионности можно использовать данные сходимости вместе с набором данных, полученных при выполнении методики в других условиях.
Все
полученные результаты (Zi)
должны принадлежать одной и той же
генеральной совокупности. Поэтому для
них рассчитывают объединенное среднее
значение
 ,
стандартное отклонение Sz
(%) и относительный доверительный интервал
,
стандартное отклонение Sz
(%) и относительный доверительный интервал
 (%)
[17]. Величина
не должна превосходить max
(%)
[17]. Величина
не должна превосходить max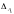 ,
т.е.:
,
т.е.:
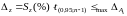 ,
(2.45)
,
(2.45)
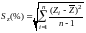 ,
(2.46)
,
(2.46)
где Sz – относительное стандартное отклонение, выраженное в процентах, рассчитанное для отношений «найдено : введено» для всех растворов; t – односторонний коэффициент Стьюдента для вероятности 95% и числа степеней свободы п–1.
Для проверки приемлемости результатов в условиях промежуточной прецизионности можно использовать также предел промежуточной прецизионности, установленный в МИ, с которым сравнивают разность средних арифметических двух серий результатов, полученных в условиях промежуточной прецизионности (СТБ ИСО 5725-6).
Для проверки приемлемости результатов, полученных в условиях промежуточной прецизионности, могут использоваться также и другие статистические оценки или проверки гипотез (например F и t критерии) [16].
По
полученным в разных условиях (серия 1 и
серия 2) результатам измерений (n1
и n2)
рассчитывают
средние арифметические ( )
двух серий измерений по формулам:
)
двух серий измерений по формулам:
 ,
(2.47)
,
(2.47)
 .
(2.48)
.
(2.48)
Рассчитывают выборочные дисперсии двух серий:
 ,
(2.49)
,
(2.49)
 .
(2.50)
.
(2.50)
Проводят оценку однородности дисперсий по критерию Фишера:
 .
(2.51)
.
(2.51)
В числителе должна находиться большая дисперсия.
Различие дисперсий незначимо (случайно) если выполняется условие:
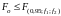 .
(2.52)
.
(2.52)
Степень свободы определяется по формулам:
 ,
(2.53)
,
(2.53)
 .
(2.54)
.
(2.54)
Сравнение выборочных средних значений по критерию Стьюдента проводят по формуле:
 .
(2.55)
.
(2.55)
Различия
между средними значениями случайны,
если выполняются условия:
 ,
где
,
где
 .
.
Воспроизводимость.
Воспроизводимость характеризует прецизионность в межлабораторном эксперименте (при совместных исследованиях).
Воспроизводимость оценивается согласно СТБ ИСО 5725-2 путем проведения независимых межлабораторных исследований на отдельных идентичных образцах (пробах), отобранных из одной и той же серии [21]. Межлабораторная воспроизводимость определяется в случае аттестации МИ, например, при включении МИ в фармакопеи.
Предел обнаружения.
Предел обнаружения соответствует минимальному аналитическому сигналу, значимо превышающему сигнал шума.
В зависимости от того, является ли методика инструментальной или неинструментальной, возможны различные подходы при определении предела обнаружения
Визуальная оценка предела обнаружения. Визуальная оценка может использоваться как для неинструментальных, так и для инструментальных методов.
Предел обнаружения устанавливают путем анализа образцов с известными концентрациями определяемого компонента и определения того минимального содержания, при котором он может надежно обнаруживаться.
Оценка предела обнаружения по соотношению сигнал/шум. Этот подход применим только к тем методам измерений, для которых наблюдается шум базовой линии. Определение соотношения сигнал/шум проводится путем сравнения измеряемых сигналов от образцов с известными низкими концентрациями и контрольных образцов. На основании полученных данных устанавливают минимальную концентрацию, при которой определяемый компонент может быть надежно обнаружен. Для оценки предела обнаружения приемлемой считается величина отношения сигнал/шум от 3:1 до 2:1.
Оценка предела обнаружения по стандартному отклонению сигнала и наклону калибровочной прямой [17]. Предел обнаружения (ПО) может быть выражен как:
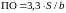 ,
(2.56)
,
(2.56)
гдe S – стандартное отклонение сигнала; b − тангенс угла наклона калибровочной прямой.
Оценка стандартного отклонения S может быть осуществлена несколькими способами:
− по стандартному отклонению для контрольного опыта. Измеряют величину аналитического сигнала для необходимого количества контрольных образцов, не содержащих определяемого компонента, и вычисляют стандартное отклонение;
− по калибровочной прямой. Необходимо проанализировать специальную калибровочную прямую, построенную для образцов с содержанием определяемого компонента, близким к пределу обнаружения. В качестве стандартного отклонения может быть использовано остаточное стандартное отклонение регрессионной прямой или стандартное отклонение точки пересечения с осью У (стандартное отклонение свободного члена линейной регрессии).
Значение предела обнаружения представляют с указанием метода, использованного для его определения. Значение предела обнаружения обычно выражается в значениях концентрации определяемого компонента в образце (пробе), например в мкг/мл. Если определение предела обнаружения основывается на визуальной оценке или оценке соотношения сигнал/шум, то представление соответствующих хроматограмм считается достаточным для его обоснования.
Если значение предела обнаружения получено путем вычислений или экстраполяции, то эта оценка должна быть подтверждена посредством независимого испытания требуемого количества образцов с содержанием определяемого компонента на пределе обнаружения или в близком к нему диапазоне.
Могут быть приемлемы и другие подходы, отличающиеся от перечисленных выше.
Ориентировочный перечень вопросов, на которые должны быть даны исчерпывающие ответы при определении предела обнаружения:
− какое минимальное содержание определяемого компонента в пробе можно обнаружить качественно;
− на основании какого метода минимальное содержание обнаружено.
Предел количественного определения.
Предел количественного определения – характеристика МИ количественного определения соединений, содержание которых в образцах (пробах) является очень низким. Предел количественного определения используется в основном для определения примесей и/или продуктов разложения.
Для установления предела количественного определения возможны различные подходы.
Визуальная оценка предела количественного определения. Визуальная оценка может использоваться как для неинструментальных методов, так и для инструментальных.
Предел количественного определения обычно устанавливают путем анализа образцов с известными концентрациями определяемого компонента и оценкой того минимального содержания, при котором определяемый компонент может быть количественно определен с приемлемой правильностью и прецизионностью.
Оценка предела количественного определения по соотношению сигнал/шум. Этот подход применим только к тем методам измерений, для которых наблюдается шум базовой линии. Определение соотношения сигнал/шум проводится путем сравнения измеряемых сигналов от образцов с известными низкими концентрациями и контрольных образцов. На основании полученных данных устанавливают минимальную концентрацию, при которой определяемый компонент может быть надежно определен количественно. Обычное соотношение сигнал/шум составляет 10:1.
Оценка предела количественного определения по стандартному отклонению сигнала и наклону калибровочной прямой [17].
Предел количественного определения (ПКО) может быть выражен как:
 ,
(2.57)
,
(2.57)
гдe S – стандартное отклонение сигнала; b − тангенс угла наклона калибровочной прямой. Значение тангенса угла наклона калибровочной прямой b может быть определено из калибровочной прямой для определяемого компонента. Оценка стандартного отклонения S может быть осуществлена несколькими способами:
− по стандартному отклонению для контрольного опыта;
Стандартное отклонение для контрольного опыта получают при измерении величины аналитического сигнала для требуемого количества контрольных образцов и вычисляют стандартное отклонение;
− по калибровочной прямой.
Следует проанализировать специальную калибровочную прямую, построенную для образцов с содержанием определяемого компонента, близким к пределу количественного определения. В качестве стандартного отклонения может быть использовано остаточное стандартное отклонение регрессионной прямой или стандартное отклонение точки пересечения с осью У (стандартное отклонение свободного члена линейной регрессии).
Значение предела количественного определения представляют с указанием метода, использованного для его определения.
Значение предела количественного определения должно быть впоследствии подтверждено анализом требуемого количества образцов с содержанием определяемого компонента на пределе количественного определения или в близком к нему диапазоне.
Ориентировочный перечень вопросов, на которые должны быть даны исчерпывающие ответы при определении предела количественного определения:
− каково минимальное содержание определяемого компонента в образце, которое можно определить количественно;
− на основании какого метода произведено количественное определение.
В случае контроля примесей нахождение величин ПО и ПКО является обязательным. При проведении валидации методик КО, данные величины не требуются, но они полезны как информация о том, насколько диапазон применения методики превосходит ее предельные возможности («запас прочности» методики).
Использование для расчетов ПО и ПКО характеристик линейной зависимости гораздо надежнее и корректнее применения отношения сигнал/шум, поскольку учитывает не только шум, но и неопределенность пробоподготовки, которая в некоторых случаях может быть определяющей.
Если линейная зависимость строилась в нормализованных координатах (т.е. ), то величины ПО и ПКО находятся в процентах к концентрации раствора сравнения, что позволяет легко оценить «запас прочности» методики.
В случае предельных тестов ПО должен быть незначим по сравнению с предельным нормируемым значением содержания примеси ImL. Для количественных испытаний незначимым по сравнению с ImL должен быть ПКО. Переходя в нормализованные координаты (в процентах к ImL) и учитывая принцип незначимости, получим [17]:
Предельные тесты:
 ,
(2.58)
,
(2.58)
Количественные тесты:
 .
(2.59)
.
(2.59)
В этом случае величины ПО и ПКО значимо не влияют на принятие решений о качестве. Из соотношений (2.58, 2.59) следует важный вывод: при контроле качества ЛС (в том числе и при контроле примесей) сами величины ПО и ПКО интереса не представляют; важно, чтобы они не превышали критические значения.
Диапазон применения.
Внутри заданного диапазона применения методики должны быть доказаны линейность, правильность и прецизионность.
Диапазон применения зависит от назначения МИ и определяется при изучении линейности.
Диапазон применения устанавливается подтверждением того, что МИ имеет приемлемую степень линейности, правильности и прецизионности при анализе образцов (проб) с содержанием (количеством) определяемого компонента в диапазоне применения МИ или на его наибольшем и наименьшем значениях.
Ниже приведены диапазоны применения МИ, которые рекомендуется рассматривать как минимально допустимые [14]:
− для количественного определения фармацевтической субстанции или лекарственного средства: от 80 % до 120 % заданной концентрации;
− для однородности дозирования: от 70% до 130% заданной концентрации, если для лекарственного средства не обоснован более широкий диапазон;
− для испытаний на растворимость, распадаемость: ±20% от значения, регламентированного ТНПА;
− для определения примесей: от значения концентрации, в которой примесь обычно обнаруживается, до 120% от регламентированного содержания. Для примесей, обладающих чрезвычайно сильным действием или имеющих токсический или непредвиденный фармакологический эффект, предел обнаружения и предел количественного определения по должны соответствовать тому уровню, на котором эти примеси должны контролироваться. При проведении валидации МИ на содержание примесей необходимо в процессе выполнения работы рассмотреть диапазон применения, внутри которого находится предполагаемый предел регламентирования примесей;
− при совместном испытании на примеси и количественном определении с использованием только образца с максимальной степенью чистоты диапазон применения должен быть линейным от обнаруживаемой концентрации примесей до 120% от указанного содержания для количественного определения.
Ориентировочный перечень вопросов, на которые должны быть даны исчерпывающие ответы при определении диапазона применения:
− какова область надежных данных по данной МИ;
− входят ли в данный диапазон измерений пробы, содержащие неизвестные концентрации определяемого компонента (например, нестабильные пробы или образцы, подвергнутые стрессовым испытаниям);
− является ли приемлемой линейность, правильность и прецизионность измерений по данной МИ в пределах от 80 % до 120% от испытуемой концентрации;
− были ли использованы по меньшей мере пять концентраций для доказательства линейности.
Требования к неопределенности результатов анализа.
Полная неопределенность результата анализа (ΔAs), в процентах, выраженная как односторонний относительный доверительный интервал для уровня доверительной вероятности 0,95 [17], не должна превышать следующих величин (табл. 2.3).
Таблица 2.3