

9.5. Загальні кількісні характеристики якості вод
Визначення загальних фізичних характеристик води
До фізичних показників води слід віднести колір, запах, каламутність, швидкість осідання завису, густину. Деякі з них можна оцінювати органолептично, а деякі вимірювати.
Колір природної води зумовлений підвищеним вмістом феруму – коричневий, глинистих речовин – жовтуватий, водоростей та інших мікроорганізмів – зеленуватий, червонястий. Колір стічної води вказує на присутність у ній певних речовин. Для визначення кольору воду не консервують, а відстоюють 2 год, потім фільтрують. Вимірюють оптичну густину води відносно дистильованої при різних довжинах хвилі при товщині шару 10 см. Колір води є доповнюючим до кольору світла, яке максимально поглинається. Можна визначити колір в градусах за дихроматно-кобальтовою шкалою, яку отримують, змішуючи у різних пропорціях ці речовини.
Запах. Якісне визначення запаху води проводять при кімнатній температурі, нагріваючи її до 50-65С у посуді, закритому годинниковим склом. Випробування проводять у приміщенні без сторонніх запахів. Запах визначають описово:
землистий – запах вологого ґрунту; торф’яний, трав’яний; затхлий, болотний; гнильний, запах цвілі; запах сіна, що гниє; ароматний; |
хлорний - запах вільного хлору; сульфідний; фенольний; запах нафти; аптечний; рибний. |
Інтенсивність запаху оцінюють у п’ятибальній шкалі: 0 – без запаху, 1, 2– дуже слабкий і слабкий, 3 – помітний, 4 – виразний, 5 – дуже сильний. Запах води спочатку оцінюють органолептично, а потім воду розбавляють декілька разів до тих пір, поки запах зникне. Визначають порогову концентрацію запаху – найменшу концентрацію (найбільше розбавляння), при якій запах ще відчутний.
Визначення грубодисперсних домішок. Природні та стічні води містять суспензію з частинок різного ступеня дисперсності. Приблизно можна визначити вміст цих домішок візуальними спостереженнями. Вода може бути:
опалесціююча – каламутна у відбитому світлі;
каламутна – непрозора у відбитому світлі та у світлі, яке пройшло;
з великими зависями – візуально помітні частинки, які при відстоюванні через 5-6 год випадають в осад.
Каламутність води можна оцінити турбідиметрично, використовуючи як стандартні розчини глинисту суспензію, стабілізовану гексаметафосфатом натрію. Вимірювання проводять у кюветах товщиною 5 см. Так можна оцінити каламутність, якщо вміст завислих речовин не перевищує 100 мг/л.
Вміст грубодисперсних домішок визначають, фільтруючи воду через фільтрувальні тиглі. Для цього через фільтрувальний тигель, попередньо доведений до сталої маси, пропускають 100-500 мл неконсервованої та невідстояної води. Тигель із залишком сушать при 100-105С протягом 2 год і доводять до сталої маси. Так визначають вміст грубодисперсних домішок (мг/л) органічних та неорганічних речовин. Якщо використовують кварцовий або порцеляновий фільтрувальний тигель, то можна визначити окремо вміст неорганічних речовин у домішках, для чого тигель після висушування при 100С прожарюють при 600С протягом 10-15 хв. За різницею визначають масу неорганічних речовин, оскільки органічні повинні при цій температурі розкластися.
Використовуючи градуйовані циліндричні ємності, у які заливають добре перемішану порцію води з грубодисперсними домішками, можна встановити швидкість осідання завислих частинок. Слідкують за висотою підняття осаду від дна через однакові проміжки часу, наприклад через 5 хв.
Густину води визначають пікнометром при сталій температурі.
Окисний потенціал води – це важливий показник, який вказує на переважання у воді окисних чи відновних процесів. Величина окисного потенціалу найбільше залежить від вмісту у ній О2 як окисника та відновників, серед яких переважно H2S. Тому визначення необхідно провести відразу після відбору проби. Якщо вода має достатньо кисню, тобто окисне середовище, то її потенціал досягає +700 мВ. В умовах відновного середовища, в якому проходить розклад органічних речовин без окиснення і накопичуються відновники, особливо сульфіди, значення потенціалу понижується до –100 мВ.
Вимірювання проводять без доступу повітря (кисню). Особливо це стосується вод з великим вмістом відновників. Вимірювання проводять потенціометром за шкалою у mV з платиновим електродом. Спочатку воду бажано пропустити через комірку 5 хв для усунення з неї повітря і кисню. Проводять заміри потенціалу, а потім повторно через 10 і 20 хв. Одержані результати повинні співпадати. Якщо відхилення потенціалу становить 5 мВ, то продовжують заміри з інтервалом 10 хв, поки не встановиться рівновага. На вимірювання окисного потенціалу води негативно впливають поверхнево-активні речовини та Mn2+, який перетворюється у MnO(OH)2.
Визначення вмісту солей у воді (мінералізації води)
Під мінералізацією води розуміють вміст розчинених неорганічних речовин. Спочатку визначають розчинені речовини. Для цього дві порції води по 50-250 мл фільтрують, поміщають у порцелянові або кварцові чашки та випаровують насухо. Чашки із залишком висушують до сталої маси при 100-105С. Маса залишку вказує на вміст у воді в розчинному стані неорганічних та стійких при 100С органічних речовин (сухий залишок). Другу порцію води після випаровування при 105С прожарюють при 600С, щоб добитися руйнування органічних речовин. Маса залишку відповідає вмісту лише неорганічних речовин, отже, мінералізації води.
Мінералізацію можна визначати також фізико-хімічними методами.
За електропровідністю знаходять вміст електролітів у воді. Для цього кондуктометрично визначають питому електропровідність води при 25С в умовах термостатування. Проводять аналіз не пізніше як через добу після відбору (розділ 10.3).
Визначення не проводять, якщо у воді є поверхнево-активні речовини.
Методом іонообмінної хроматографії знаходять сумарний вміст солей у воді. Воду пропускають через сильноосновний катіоніт у Н+-формі. Катіони макроелементів з води обмінюються на кислоту, яку визначають кислотно-основним титруванням.
Спочатку через колонку з катіонітом пропускають розчин 0,100 М HCl, після чого її надлишок вимивають водою.
Відбирають 10,00 мл води і пропускають через підготовлену колонку зі швидкістю 1 крапля/с. Розчин, який витікає, збирають у колбу для титрування. Для повного вимивання кислоти через колонку додатково пропускають 50-80 мл дистильованої води і збирають в ту саму колбу. Одержаний розчин кислоти титрують 0,100 М стандартизованим розчином NaOH з індикатором фенолфталеїном.
Вираховують загальний вміст солей у воді:
С = CE(NaOH)V(NaOH)100 (ммоль/л).
Визначення загального вмісту органічних речовин у воді за окиснюваністю (хімічного споживання кисню ХСК)
Окиснюваність води (ХСК) – це кількість сильного окисника, який окиснює органічні речовини в одиниці об’єму води. Кількість окисника є еквівалентною до кількості кисню, який міг би окиснити речовини. Переважно під окиснюваністю розуміють загальний вміст розчинених органічних речовин (див. розділ 4.2). Вода вважається питною, якщо ХСК<5 мг О/л, придатною для господарських цілей при ХСК<30 мг О/л. Елементи C, H, S, P та інші в органічній речовині дією сильного окисника окиснюються до СО2, Н2О, Р2О5, SO3, а нітроген у кислому середовищі утворює NH4+.
Методи, які використовуються для визначення ХСК, дають результати, близькі до теоретичного значення, однак можливі відхилення. У процесі визначення ХСК разом з органічними сполуками окиснюються неорганічні відновники. Їх вміст попередньо визначають і віднімають від знайденого значення ХСК.
Розрізняють такі методи залежно від застосовуваного окисника: перманганатний, йодатний, дихроматний та церієвий. Максимальне окиснення органічних речовин у воді досягається, якщо застосовувати дихроматний та йодатний методи у присутності концентрованої сульфатної кислоти.
Для аналізу питних вод, які містять <300 мгСl-/л, застосовують перманганатний метод з йодометричним закінченням. Недоліки методу – у процесі кип’ятіння розчинів надлишок перманганату розкладається:
2HMnO4 = H2MnO4 + MnO2 + O2.
Визначення ХСК треба проводити у свіжовідібраних пробах. Якщо це неможливо і аналіз буде проведений не пізніше, ніж через 48 год, то пробу консервують, додаючи розведену (1:2) H2SO4.
Перш ніж визначати ХСК, воду фільтрують через мембранний фільт (паперовий попередньо промивають гарячою водою). Якщо фільтрування недопустиме (у воді є речовини, які легко окиснюються на повітрі), то воду тривалий час відстоюють, а для аналізу відбирають верхній шар.
Дихроматний арбітражний метод визначення ХСК. Органічні речовини окиснюють дихроматом:
C6H12O6 + 4Cr2O72- + 32H+ = 6CO2 + 22H2O + 8Cr3+,
надлишок якого відтитровують розчином солі Fe(ІІ) за наявності фероїну як індикатора, або потенціометрично:
Cr2O72-+6Fe2++14H+ 2Cr3++6Fe3++7H2O.
Окиснення прискорюється та охоплює майже всі органічні сполуки, якщо використовувати як каталізатор Ag2SO4.
Суміш води, окисника, концентрованої сульфатної кислоти та каталізатора вносять у колбу, закривають корком зi зворотним холодильником та нагрівають 2 год при слабкому кипінні. Після охолодження суміш кількісно переносять у конічну колбу, розбавляють дистильованою водою, додають індикатор, а тоді відтитровують надлишок калій біхромату розчином солі Мора. Одночасно виконують сліпу пробу з 50,00 мл дистильованої води.
Результат визначення ХСК становить 95-98% від теоретичного. В деяких органічних речовинах нітроген окиснюється з утворенням N2 замість NH4+, що веде до помилки визначення ХСК. Дихроматом не окиснюються або повільно окиснюються піридин і його гомологи (пірол, піролідин), нікотинова кислота, інші нітрогеновмісні вуглеводні, парафін і нафталін. Сильно забруднену воду перед окисненням розводять.
Якщо проба містить неорганічні відновники, то їх кількість визначають окремо іншими методами і вміст віднімають від результату визначення ХСК. При цьому не враховують вмісту H2S та SO2, оскільки під час додавання H2SO4 вони видаляються. Хлорид-іони окиснюються до хлору, тому хлорид маскують меркурій (ІІ) сульфатом з утворенням малодисоційованого [HgCl4]2-. У випадку великої кількості хлоридів та органічних сульфідів до проби води спочатку додають дихромат, потім меркурій (ІІ) сульфат, щоб утворився осад HgS, а лише тоді сульфатну кислоту та кип’ятять.
Визначенню заважають іони NO2-. Для їх видалення додають сульфанілову кислоту і пробу кип’ятять:
H2NSO2OH+HNO2 N2+H2SO4+H2O;
H2NSO2OH+H2O NH4HSO4.
Величину ХСК, як число мг О/л, обчислюють за формулою:
 ,
мг О/л.
,
мг О/л.
Величина проби води, яку аналізують, різна. Якщо ХСК води є в межах 50-500 мгО/л, то на аналіз беруть 10,0 мл проби; в межах 500-4000 мг О/л, то відбирають 1,0 мл проби і розбавляють дистильованою водою; якщо понад 4000 мг О/л, то пробу розбавляють. У випадку, коли ХСК води менше за 50 мг О/л, цей метод застосовувати не можна.
Прискорений дихроматний метод визначення ХСК використовують для серійного аналізу з метою контролю за станом води у водоймі. Результати визначення ХСК можуть бути дещо заниженими, особливо за наявності речовин, які важко окиснюються (ацетатна кислота, бензол та ін.). Особливість методу полягає у використанні великого надлишку концентрованої сульфатної кислоти (7,5 мл на 1,00 мл проби води), яка під час розчинення підвищує температуру розчину до 100°С. Через 2 хв охолоджують розчин до кімнатної температури, додають 100 мл дистильованої води і титрують надлишок дихромату розчином солі Fe (ІІ).
Перманганатний метод (Кубеля) визначення ХСК ґрунтується на окисненні речовин перманганатом у сульфатно-кислому середовищі. Застосовується для аналізу питної та поверхневих слабко забруднених вод, які містять до 300 мг/л Cl-.
Для визначення залишкової концентрації KMnO4 в ту ж колбу до гарячого розчину доливають 0,005 М розчин H2C2O4 і титрують 0,00200 М розчином KMnO4 до рожевого забарвлення, яке не зникає протягом 1 хв. Обчислюють окиснюваність:
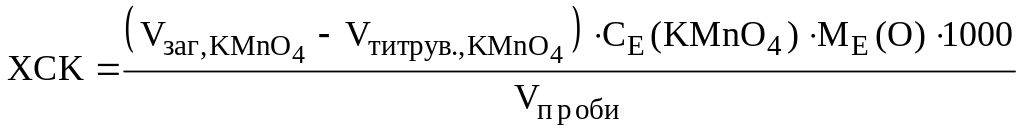 мг О/л.
мг О/л.
Заважають визначенню відновники: Cl-, S2-, NO2-, Fe(II).
Фотометричне визначення перманганатної окиснюваності питної води. Оскільки вміст органічних речовин у природних водах можна вимірити в УФ діапазоні електромагнітного випромінювання при =254 нм, то орієнтовну величину перманганатного окиснення (ПО) води можна оцінити за вимірюванням оптичної густини води при цій довжині хвилі за формулою: ХСК(ПО)= 27,7А254 (Хоффманн М., Галаган А.А., 1999).
Співвідношення ХСК / m(Сорг.) вказує орієнтовно на тип органічних речовин, які є у воді:
![]() .
.
Це співвідношення має найменше значення для Н2С2О4, вважаючи, що окисником є кисень:
3H2C2O4+Cr2O72-+4H+ = 6CO2+2Cr3++7H2O
або 2Н2С2О4+О2 = 2Н2О+4СО2, тоді
 .
.
Якщо співвідношення ХСК/m(Сорг.) більше за 4,1, то у воді в основному знаходяться похідні метану. Наприклад, для окиснення метану:
СH4+2О2=2Н2О+СО2,
![]() .
.
При співвідношенні, більшому за 3, вода забруднена речовинами з великим вуглецевим ланцюгом, при 2,7-2,8 – речовинами білкової природи, при 2,67 – вуглеводами.
Визначення розчиненого у воді кисню
Вміст кисню у воді має значення для оцінки її корозійних властивостей, придатності для рибних господарств (4–6 мг/л). У дистильованій воді при температурі 20С вміст розчиненого кисню становить 9 мг/л, у поверхневих водах – 6-10 мг/л, а в період найбільш інтенсивного фотосинтезу досягає 16 мг/л. Визначають кількість розчиненого кисню у воді, яка була піддана хімічній та біохімічній очистці, а також як один з етапів аналізу при визначенні біохімічного споживання кисню. Визначення необхідно проводити відразу після відбору проби води.
Методом Вінклера визначають кисень при його орієнтовному вмісті, не меншому, ніж 0,2-0,3 мг/л. Якщо у воді є відновники, то визначення проводять модифікованим методом Росса.
Суть визначення полягає в окисненні розчиненим у воді киснем манган (ІІ) гідроксиду до манган (ІV) гідроксиду з наступним його відновленням йодидом. В основі лежать такі реакції:
2Mn(OH)2 +О2 2MnO(ОН)2,
MnO(ОН)2+ 4H+ + 2J- Mn2+ + J2 + 3H2O,
Виділений йод титрують стандартизованим розчином тіосульфату:
J2+ 2S2O32- 2J-+S4O62-.
За наявності у воді відновників (S2-, SO32-, NO2-) їх попередньо окиснюють натрій гіпохлоритом, надлишок якого відновлюють калій тіоціанатом. Якщо вода містить активний хлор, то його теж видаляють додаванням калій тіоціанату. Відбуваються такі реакції:
Red+ClO-+ 2H+ Ox+Сl-+ Н2O,
4СlO- +SCN-+2OH- OCN- +4Сl- +SO42- +H2O.
Після цього аналіз завершують за методом Вінклера.
Перед аналізом пробу води певним чином підготовляють.
Воду перед визначенням не фільтрують, а лише відстоюють.
Якщо важкорозчинні зависі погано відділяються відстоюванням, то пробу води освітлюють додаванням Al(OH)3 (на 1 л води - 10 мл 10% розчину KAl(SO4)312H2O та 2 мл концентрованого розчину NH3).
У присутності зависів та мікроорганізмів, здатних засвоювати кисень, до води, крім Al(OH)3, додають дезінфікуючі речовини: сульфамінову кислоту або меркурій (ІІ) хлорид (на 1 л води – 10 мл суміші NH2SO3H з HgCl2).
Якщо зависі добре відділяються при відстоюванні, однак вода містить мікроорганізми, то додають лише дезінфікуючі речовини (10 мл суміші сульфамінової кислоти та мідного купоросу (45 мл 0,67 М розчину NH2SO3H змiшують з 45 мл 0,4 М СuSO45H2O та 2,5 мл концентрованої СH3COOH i доводять водою до 100 мл).
Пробу води зберігають у посуді так, щоб під корком не залишилось повітря.
Іони Fe(ІІІ) зв’язують фторидом (на 1 л води перед розчиненням осаду гідрооксидів кислотою додають 1 мл 20% розчину NaF), або фосфатом (осад гідрооксидів розчиняють у 10 мл 25% Н3РО4).
NO2- видаляють сечовиною.
У склянки з водою піпеткою вводять точно відомий об’єм розчину солі мангану, потім іншою піпеткою розчину КОН. Після випадання осаду в склянку вводять піпеткою 10,0 мл розчину H2SO4 (1:4) або 25% розчину H3PO4 i 0,3 мл 20% розчину сечовини. Закриту склянку перемішують, після чого воду кількісно переносять у колбу та приливають 2 мл розчину калій йодиду. Через 5 хв виділений йод відтитровують розчином натрій тіосульфату з крохмалем. Аналізують три-п’ять паралельних проб. Обчислюють вміст розчиненого у воді кисню:
![]()
де V1 – об’єм склянки, мг/л;
V2 – сумарний об’єм доданих реактивів, якими зв’язували кисень.
Полярографічне визначення кисню ґрунтується на вимірюванні висоти хвилі відновлення його:
О2+2![]() +2Н+Н2О2,
+2Н+Н2О2,
Н2О2+2 +2Н+2Н2О.
Величина потенціалу залежить від рН. Для аналізу застосовують першу хвилю на фоні насиченого NaCl або 70% NaOH. При визначенні заважають інші деполяризатори. Щоб усунути їх вплив, спочатку визначають висоту хвилі у пробі води, а потім через воду пропускають інертний газ для видалення кисню і знову одержують хвилю. Різниця висот цих двох хвиль відновлення відповідає вмісту лише кисню. Для побудови градуйованого графіка готують дистильовану воду з точно відомим вмістом О2.
Визначення біохімічного споживання кисню (БСК)
Біохімічним споживанням кисню (БСК) називають кількість О2, який повинен затратитися на окиснення в 1 л води органічних речовин за участю аеробних мікроорганізмів, однак без затрати кисню на процес нітрифікації. Це кількість кисню, спожитого мікроорганізмами у процесі життєдіяльності. Величина біохімічного споживання кисню є менша за величину хімічного споживання кисню.
Стандартно встановлення величини біохімічного споживання кисню триває 5 діб при 20С без доступу світла і повітря. Повне біохімічне споживання кисню визначають протягом 20 діб, але може бути і менше. Фактично визначення припиняють у момент появи у пробі води NO2- в концентрації 0,1 мг/л. Можна визначати БСК за умови пригнічення діяльності нітрифікуючих бактерій, тоді біохімічно окиснюються лише органічні речовини.
Визначення БСК полягає у контролі вмісту розчиненого кисню у воді протягом всього експерименту. Загалом протягом 5 діб вміст кисню може понизитися на 2 мг/л, а залишковий вміст повинен становити 3 мг/л. Це відповідає вимозі протікання аеробних процесів протягом всього експерименту.
Перед визначенням пробу води підготовляють.
Якщо величина БСК надто велика, то проби води розбавляють.
Воду з завислими частинками відстоюють півгодини або з неї відфільтровують зависі.
Якщо вода надто кисла або лужна, то її нейтралізують.
У випадку хлорованої води усувають „активний хлор” або відстоюючи воду протягом 2 год, або відновлюючи сполуки хлору дією Na2SO3, причому необхідну кількість відновника перед тим визначають йодометрично.
Повне БСК можна вважати як різницю ХСК на початку дії на пробу мікроорганізмів та в кінці дії (у присутності інгібіторів бактерій-нітрифікаторів). Визначають окремо ХСК води і окремо ХСК твердої фази або добре перемішаної твердої фази (зависів) з водою.
Для визначення БСК проби води поміщають у 6 скляних колб, закритих притертими корками так, щоб під корком не залишалось повітря. У 6 таких самих колб вводять дистильовану воду. Спеціальні колби з ковпачками, у які залита вода для захисту від контакту з повітрям, показані на рис.9.3.
У першій порції води відразу визначають вміст розчиненого кисню (хлорованої води – через 2 год), також визначають вміст кисню у першій порції дистильованої води.
Усі проби витримують при 20С без доступу повітря.
Наступні проби аналізують через 2, 3, 5, 10, 20 діб, одночасно визначаючи у ній вміст нітриту. Якщо через певний відрізок часу з’явився нітрит у концентрації 0,1 мг/л, то припиняють витримування проб води, тобто процедуру визначення БСК.
|
Рис.9.3. Колба для інкубації проби води, застосовувана при визначенні біохімічного споживання кисню |
Обчислюють БСК проб нерозведеної води:
![]()
Якщо пробу води брали невелику (V мл) і розбавляли, то обчислення ведуть за формулою:
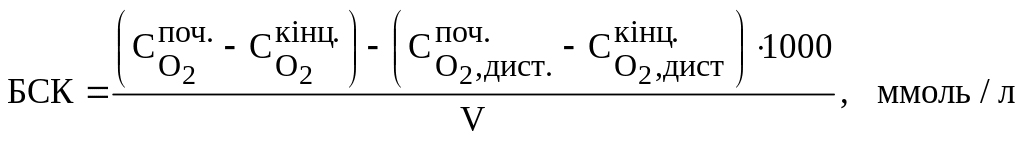 .
.
Показники, визначені з величин ХСК та БСК
За величиною БСК та ХСК рідкої та окремо відділеної твердої фази визначають низку важливих показників:
частка
органічних речовин, які не окиснюються,
 ;
;
частка, яка припадає на утворену при споживанні кисню біомасу,
 ;
;
частка
біохімічно окиснюваних речовин,
![]() ;
;
кількість речовин, які не окиснюються біохімічно на момент часу t,
![]() .
.![]()
Визначення хлороємності води
Хлороємність води визначають для встановлення кількості „активного хлору”, необхідного для дезінфекції води. Сполуки хлору у вищих ступенях окиснення окиснюють органічні речовини, серед яких мікроорганізми та неорганічні відновники. Якщо у воду потрапили ціаніди, то їх можна окиснити лише, якщо рН>10:
Cl2+CN-+2OH-= 2Cl-+OCN-+H2O.
При меншому значенні рН утворюється ClCN, який веде себе як активний хлор і спотворює результати визначення:
Cl2+2CN-= 2ClCN, ClCN+Red = Cl-+CN-+Ox.
Знезараження води хлоровмісними окисниками описано в розділі 6.3.
Визначення хлороємності проводять у відфільтрованій, відстояній воді або у воді з осадом йодометричним методом у двох серіях проб води. Однакові проби води відбирають у ряд посудин, у кожну з яких додають відому і щоразу більшу кількість хлорної води або хлорного вапна (умовно Cl2). Після відстоювання залишок Cl2 відтитровують йодометрично.
У першій серії після додавання хлору проби відстоюють 5 хв і визначають речовини, які швидко окиснюються, а в другій серії відстоюють 1-2 год, що відповідає режиму хлорування води. Після відстоювання до проб води додають розчин KJ і титрують виділений J2 тіосульфатом з індикатором крохмалем.
У склянці відбувається процес: Cl2+Red = 2Cl-+Ox,
надлишок Cl2+2J-= J2+2Cl-.
Титрування: J2 + 2S2O32- 2J- + S4O62-.
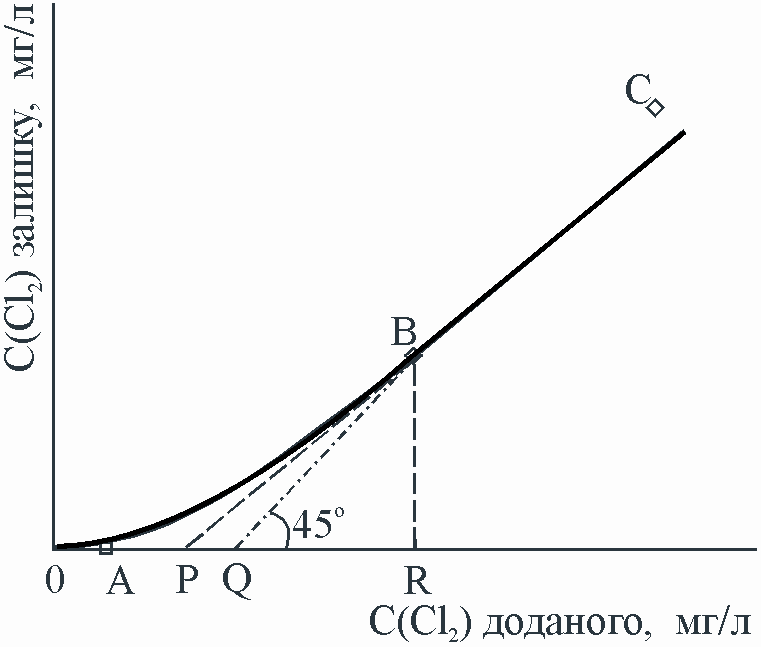
За результатами титрування будують для кожної серії залежність вмісту Cl2, який залишився, від вмісту доданого Cl2 (рис.9.4).
Якщо у воді є амоній та аміни, то на них затрачається зайва кількість хлору і виділяється зайва кількість йоду:
Cl2+RNH2 = RNHCl+HCl і RNHCl+2HJ = RNH2+J2+HCl.
Тому початкова ділянка (0В) набирає іншого вигляду (рис.9.5).
Для визначення повної хлороємності з точки В на графіку проводять перпендикуляр на вісь абсцис (точка R) і пряму під кутом 45 до перетину з віссю абсцис. Відрізок 0R відповідає повній затраті сполук хлору. Відрізок 0Q відповідає затраті хлору на окиснення всіх відновників, окиснення води – це повне хлорування води. Відрізок 0P відповідає затраті хлору на окиснення лише органічних речовин – істинна хлороємність води.
|
Рис.9.4. Крива для визначення хлороємності води: 0А – речовини, які швидко окиснює Cl2; АВ – речовини, які повільно окиснюються; ВС – окиснення припинилось; PВ – екстраполяція відрізка ВС (припинення окиснення); PQ–окиснення води хлором (Cl2+H2O=HCl+HClO, 2HClO=2HCl + O2); BP – екстраполяція реального ходу вітки ВC |
Утворені хлораміни при подальшому додаванні хлору руйнуються:
2NH3 + 3Cl2 = N2 + 6HCl,
2CH3–NH2 + 3Cl2 = N2 + 4HCl + 2CH3–Cl,
крива проходить через максимум і дальший хід графічної залежності відповідає ділянці ВС, як на рис. 9.4.
|
Рис.9.5. Крива визначення хлороємності води за умови присутності амонію та амінів |
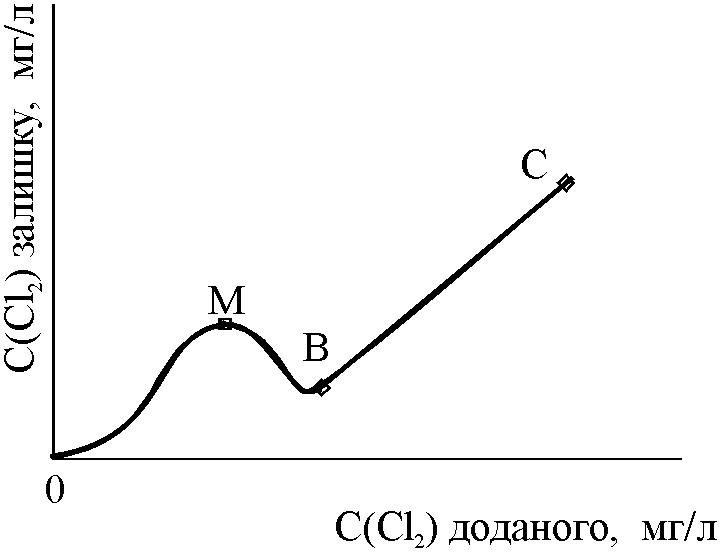
Питні води хлорують до появи хлорамінів (точка М), а стічних – до розкладу хлорамінів, використовуючи екстраполяцію прямолінійної ділянки ВС, як і у випадку кривої визначення хлороємності без амінів.
Визначення соленості води
Загальна соленість води зумовлена вмістом усіх розчинених солей (всіма аніонами: НСО3-, СО32-, ОН-, SO42-, Cl-, ...), які є у воді.
Некарбонатна соленість обумовлена аніонами, за виключенням тих, які підлужнюють воду, тобто, крім НСО3-, СО32-, ОН-.
Визначення некарбонатної соленості води. Через катіоніт у Н+-формі пропускають воду (200,0 мл) і відбувається обмін:
R-H+ + Сa2+, Mg2+, Na+, K+ nH+ + R-Сa2+,Mg2+,Na+,K+.
Однак перші порції витисненої проби (~80 мл) відкидають, оскільки вони містять продукти нейтралізації лугів та карбонатів:
НСО3-, СО32-, ОН- + Н+ Н2О+СО2.
Наступні порції води, витисненої з колонки, збирають у мірну колбу на 100,0 мл. Кількість іонів металів еквівалентна до кількості витисненої кислоти.
Через одержану пробу води пропускають повітря для видалення з
неї СО2. Тоді 100,0 мл проби, яка містить витиснену кислоту, титрують 0,100 М розчином NaOH за метилоранжем. Обчислюють некарбонатну соленість води:
 .
.
Загальна соленість води це сума некарбонатної соленості та загальної лужності, тобто вміст усіх аніонів.
Визначення твердості води
Загальну твердість води (загальний вміст солей кальцію і магнію) визначають комплексонометричним титруванням у слаболужному середовищі з індикатором хромогеном чорним, причому наявні у воді мікроелементи, через їхній малий вміст та зв’язування у комплекси, не заважають визначенню.
Якщо в стічній воді підвищений вміст важких металів, то їх маскують зв’язуванням у ціанідні комплекси або осаджують дією Na2S у вигляді важкорозчинних сульфідів, Mn2+ зв’язують дією NH2OH.
Перед титруванням забрудненої води з зависями, яка містить багато органічних речовин, природних гумусових сполук, її фільтрують. Фільтрат випаровують та прожарюють при 600С, а одержаний залишок розчиняють в кислоті, після чого титрують розчином ЕДТА.
Загальну твердість води (Тзаг.(H2O)) виражають сумарною кількістю солей кальцію і магнію, ммоль/л:
Тзаг.(H2O)=![]() ммоль/л.
ммоль/л.
Карбонатна твердість води зумовлена вмістом у ній розчинних гідрогенкарбонатів кальцію та магнію. У процесі кип’ятіння гідрогенкарбонати перетворюються в карбонати і випадають в осад:
Ca(HCO3)2![]() CaCO3¯+CO2+H2O,
CaCO3¯+CO2+H2O,
2Mg(HCO3)2 (MgОН)2CO3¯+3CO2+H2O.
Карбонатна твердість води – це, по-суті, її загальна лужність.
Вміст гідрогенкарбонатів кальцію та магнію визначають, титруючи пробу води стандартизованим розчином хлоридної кислоти за метилоранжем:
Ca(HCO3)2+2HCl CaCl2+H2O+2CO2,
Mg(HCO3)2+2HCl MgCl2+H2O+2CO2,
або HCO3- + H3O+ H2CO3 + H2O.
Обчислюють карбонатну (тимчасову) твердість води:
 ,
ммоль/л.
,
ммоль/л.
Постійна твердість води зумовлена розчинними сульфатами та хлоридами кальцію та магнію. Її вираховують як різницю між загальною та тимчасовою твердістю води.
Визначення нітритів
Надмір нітритів у воді свідчить про перебіг процесу нітрифікації, а наявність їх в концентрації, що перевищує 0,1 мг/л, свідчить про інтенсивний процес нітрифікації.
Визначення проводять якомога швидше після відбору проби, оскільки нітрити можуть як окиснитися у нітрати, так і відновитися за участю відповідно аеробних та анаеробних мікроорганізмів. При необхідності воду консервують додаванням розчину сульфатної кислоти. До води з зависями та забарвленої води додають суспензію Al(OH)3, яка сприяє коагуляції домішок.
Нітрити
визначають фотометрично
з сульфаніловою
кислотою
та -нафтіламіном
(реактив Гріса) при рН=2 у вигляді
пурпурного азобарвника (![]() =40103)
у межах концентрацій 1-0,01 мг/л:
=40103)
у межах концентрацій 1-0,01 мг/л:

Заважають реакції сильні окисники, відновники, аліфатичні аміни.
Визначення загального вмісту амінів, амідів, пептидів. Визначають сумарний вміст амінів, імінів, амінокислот, речовин з пептидними зв’язками, що містять до 10 аміногруп. Це продукти розкладу білкових речовин, присутність яких у воді вказує на забруднення нерозкладеними біологічними речовинами. Визначення проводять фотометрично з нінгідрином у вигляді фіолетової сполуки.
Нінгідрин взаємодіє також з аміаком та амонієм. Тому попередньо визначають NH3 i NH4+ за методом К’єльдаля. Заважають визначенню іони Cu2+, Cd2+, Fe3+, які є інгібіторами реакції. Їх маскують зв’язуванням у ціанідні комплекси.

Визначення сполук сульфуру
У воді сполуки сульфуру є в різних формах. Сульфати належать до макроелементів. Низка органічних речовин містять сульфур. Якщо вода забруднена продуктами розкладу органічних речовин біологічного походження, то в ній, особливо у придонних шарах, з’являються сульфіди, меркаптани, сульфіти, тіосульфати. Крім того, деякі стоки містять велику кількість сполук сульфуру в нижчих ступенях окиснення. Саме такі речовини є забруднювачами води і їхній вміст треба контролювати.
Спочатку визначають вміст іонів SO42- у воді. Визначення можна проводити різними методами.
Гравіметрично осаджують барій сульфат, осад прожарюють при 800С:
Ba2++SO42-=BaSO4.
Якщо у стічній воді є CrO42-, то він заважає визначенню. Тому Cr(VI) відновлюють при кип’ятінні у розчині хлоридної кислоти і гліцерину або етилового спирту. Утворений Cr3+ зв’язують з ЕДТА. Воду, яка містить сульфіти та тіосульфати, підкислюють і кип’ятять до повного видалення SO2.
Комплексонометричне титрування. Спочатку осаджують BaSO4. Осад розчиняють в лужному середовищі у ЕДТА при кип’ятінні:
BaSO4+H2Y2-+2OH-![]() BaY2-+2H2O
+ SO42-.
BaY2-+2H2O
+ SO42-.
Надлишок ЕДТА відтитровують стандартним розчином солі магнію.
Обчислюють вміст сульфату:
![]() ,
мг/л.
,
мг/л.
Визначення сульфуру органічних речовин. У порції води, такій же, як для визначення сульфату, в лужному середовищі окиснюють органічні речовини дією Br2 у CCl4 (при малому вмісті сполук сульфуру – без органічного розчинника). Утворюється сульфат:
CH3–SH + 7Br2 + 16OH- 14Br- + SO42- + CO2 + 10H2O.
Розчин підкислюють HNO3 та нагрівають для видалення залишку Br2. Після цього суміш переносять у платиновий тигель та випаровують у присутності Na2CO3 i MgO. Сухий залишок розчиняють у HCl та визначають вміст SO42-. За різницею визначеного загального вмісту сульфату, який включає і сульфат, утворений під час окиснення органічних речовин, та вмісту вільного сульфату, визначають вміст сульфату, утвореного з органічних речовин.
Визначення загального вмісту сульфідів (S2-, HS-, H2S). Сульфіди містяться в стічних водах, у підземних водах або в забруднених біологічними залишками придонних водах.
Йодометричним титруванням визначають великий вміст сульфідів (5-20 мг). Воду консервують додаванням розчину солі кадмію з утворенням важкорозчинного CdS. Одержаний осад розчиняють, додаючи стандартизований розчин йоду і підкислюючи хлоридною кислотою. Надлишок йоду відтитровують тіосульфатом:
CdS+ J2 = Cd2++ 2J- + S,
титрування: J2 + 2 S2O32- 2J- + S4O62-.
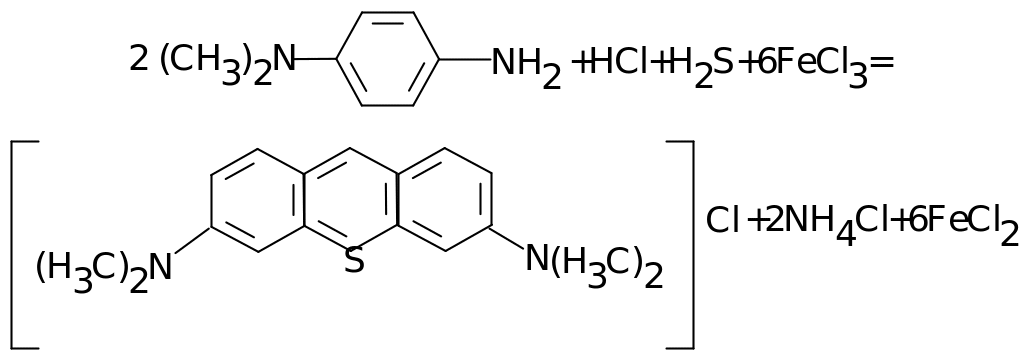
При малому вміст (у межах 0,005-1,0 мг/л) сульфід визначають фотометрично з метиленовою синню (диметил-п-фенілендиаміном) у сильнокислому середовищі. Це проба на загнивання води. На місці відбору воду консервують, вливаючи пробу води у розчин (CH3COO)2Zn. Реактив є в сильнокислому середовищі, що може спричинити виділення СО2. Тому розчин реактива заливають зверху на пробу води, закривають та збовтують. Відбувається взаємодія, яка проходить через декілька стадій. Сумарний процес можна відобразити рівнянням:
Визначення
чутливе,
![]() =34103,
заважають нітрити, тому їх руйнують
сульфаміновою кислотою, яка входить у
склад реактиву:
=34103,
заважають нітрити, тому їх руйнують
сульфаміновою кислотою, яка входить у
склад реактиву:
NH2SO2OH + HNO2 = N2 + H2SO4 + H2O.
Визначення вмісту S2-, SO32-, S2O32- проводять у трьох однакових пробах води методами редоксиметричного титрування.
Сумарне визначення S2-, SO32-, S2O32 проводять у першій пробі. Для цього пробу води вливають у підкислений ацетатною кислотою розчин стандартизованого розчину йоду, залишок якого відтитровують стандартизованим тіосульфатом з індикатором крохмалем:
S2- + J2 = S +2J-,
SO32- + J2 + H2O = SO42- +2J- + 2H+,
2S2O32- + J2 = S4O62- +2J-,
титрування: J2 + 2 S2O32 2J- + S4O62-.
Визначення S2O32-. У другу порцію води вводять 10% розчин (CH3COO)2Zn і осаджують ZnS, додають розчин формальдегіду і ацетатну кислоту і надлишок стандартизованого розчину йоду. За цих умов формальдегід зв’язує сульфіт, отже у взаємодію з йодом вступає лише S2O32-. Його вміст визначають аналогічно йодометричним титруванням.
Визначення S2-. Додаванням розчину BaCl2 осаджують сульфіт у формі BaSO3. Створюють слабколужне середовище з рН 9,2 і титрують сульфіди стандартизованим розчином K3[Fe(CN)6], причому за цих умов не вступає у реакцію S2O32-. Індикатором служить розчин Fe2+ і диметилгліоксим. Відбувається реакція:
S2- +2[Fe(CN)6]3- = S +2[Fe(CN)6]4-.
Визначенню заважають іони тіоціанату. За результатами титрування визначають вміст S2-.
Визначення сполук хлору
Сполуки хлору переважно добре розчинні у воді. Хлорид належить до макроелементів. Якщо у прісній воді вміст хлоридів невисокий, то у соляних водах перевищує вміст інших макроелементів.
Сполуки хлору у вищих ступенях окиснення можуть знаходитись у деяких стічних водах і містяться у водах, які знезаражували хлоруванням. Ці сполуки також добре розчинні у воді.
Визначення хлоридів
Велику кількість Cl- (>15 мг/л) визначають титриметрично стандартизованим розчином AgNO3 або Hg2(NO3)2:
Ag+ + Cl- = AgCl,
Hg22+ + 2Cl- = Hg2Cl2.
Якщо вода містить багато органічних речовин і забарвлена, то її попередньо мінералізують: випарюють з Na2CO3 і прожарюють, потім залишок розчиняють у кислоті. Можна осадити хлорид дією солі аргентуму, осад відфільтрувати, а надлишок Ag+ у розчині визначити титруванням тіоціанатом з індикатором Fe3+:
титрування: Ag + SCN- AgSCN,
реакція з індикатором: Fe3++ nSCN- Fe(SCN)n3-n.
Малу кількість хлориду (<3-5 мг/л) визначають фотометрично з дифенілкарбазидом у вигляді фіолетової адсорбційної сполуки з осадом Hg2Cl2 при рН 8. Великий вміст органічних речовин заважає визначенню, тому їх попередньо руйнують.
Визначення загального вмісту сполук хлору (Cl-, Cl2, ClO-, NH2Cl, NHCl2, ClO2-, ClO3-) проводять після відновлення цих сполук до хлориду. Всі сполуки-окисники хлору відновлюють сіллю Мора, що можна зобразити схемою:
Cl2 (ClO-, NH2Cl, ClO2-, ClO3-) +2Fe2+ 2Cl- +2Fe3+ + H2O.
Вміст хлориду визначають титриметрично за методом осадження. Від одержаного вмісту хлориду після відновлення віднімають вміст хлориду, виявленого уводі, і за різницею обчислюють вміст хлориду, утвореного з окиснених форм хлору.
Визначення “активного хлору” (Cl2, ClO-, NH2Cl, NHCl2) проводять або після відновлення цих сполук до хлориду, або йодометричним титруванням їх як окисників. „Активний хлор” (Cl2, ClO-, HClO, RNHCl) титрують йодометрично при рН 4,5, бо в більш кислому середовищі заважають інші окисники, зокрема ClO3-.
RNHCl+2HJ = RNH2+J2+HCl,
титрування: J2+2S2O32- 2J-+ S4O62-.
Визначення ClO-, ClO2-, ClO3-. Йодометрично, відновлюючи сполуки хлору у вищих ступенях окиснення арсенітом, можна визначити окремо сполуки хлору у різних ступенях окиснення, такі як ClO-, ClO2-, ClO3-. Проводять визначення в однакових пробах води.
Для визначення ClO- до надлишку стандартизованого арсеніту у присутності гідрогенкарбонату додають пробу, що містить окисні сполуки хлору. Суміш відстоюють, додають розчин ацетатної кислоти до початку виділення СО2, а тоді, у присутності крохмалу як індикатора, титрують надлишок арсеніту розчином йоду. Відбуваються реакції:
ClO- + AsO33- = Cl- + AsO43-,
титрування: AsO33- + J2 + H2O = AsO43-,+ 2J- + 2H+.
Визначення ClO- і ClO2- проводять аналогічно, але у розчині сильної кислоти (2M H2SO4), бо в цих умовах не заважає ClO3-. За різницею обох титрувань визначають вміст ClO2-.
Визначення ClO3- у стічних водах. Окисні сполуки хлору в присутності хлоридної кислоти окиснюють арсеніт, надлишок якого відтитровують броматом з індикатором метиловим оранжевим, який внаслідок окиснення бромом знебарвлюється. Відбуваються реакції:
3H3AsO3 + ClO3- = 3H2AsO4- + Cl- + 3H+,
а також H3AsO3 + ClO- = H2AsO4- + Cl- + H+;
титрування: 3H3AsO3 + BrO3- 3H2AsO4- + Br- + 3H+,
BrO3- + 5Br- + 6H+ = 3Br2 + 3H2O.
За результатами цього титриметричного визначення та попереднього визначення вмісту у воді ClO- і ClO2- обчислюють за різницею вміст ClO3-.
Визначення загального вмісту важких металів
Визначати загальний вміст важких металів можна лише після того, як зруйновано сполуки, у які вони можуть бути зв’язані (комплекси з неорганічними та органічними лігандами, фульвокислотами, гідроксиди та гідроксокомплекси). Для цього у пробі води кислотами обзолюють органічні речовини, а отриманий залишок розчиняють в розведеній кислоті. В одержаному розчині солей іони Fe3+, Al3+, Mg2+ зв’язують тартратом або цитратом, іони Mn2+ – гідроксиламіном.
Визначення проводять екстракційно-фотометрично. У підготовленій пробі води створюють рН 8-8,5 і додають розчин дитизону в CCl4. Катіони важких металів переходять в органічний шар у вигляді дитизонатів:
Me2++
![]() + 2OH-
+ 2OH-
![]() + 2H2O.
+ 2H2O.
З органічного розчину іони металів реекстрагують 0,01% водним розчином Hg(NO3)2. При цьому іони Hg2+ заміщають катіони важких металів з утворенням оранжевого дитизонату меркурію, який визначають фотометрично, а інші катіони переходять у водний розчин:
+
Hg2+
![]() + Me2+.
+ Me2+.
За результатами фотометрування комплексу дитизонату меркурію в органічному розчиннику визначають кількість меркурію, яка є еквівалентною до загальної кількості катіонів важких металів.