

10.2.3. Застосування вольтамперометричних методів аналізу
Вольтамперометрія це група електроаналітичних методів, які ґрунтуються на використанні поляризаційних кривих, одержаних з допомогою малого електрода, що легко поляризується і занурений в аналізований розчин.
У методі вольтамперометрії електроліз розчину аналізованої речовини проходить за умови поступового збільшення напруги, при цьому фіксують силу струму. Одержана крива залежності сили струму від напруги називається вольтамперною або поляризаційною кривою, тому цей метод також має назву полярографії. Необхідною умовою вольтамперометрії є використання електрода, який легко поляризується і має невелику поверхню. Якщо електроліз проводять без перемішування електроліту, густина струму на електроді висока (електрод має малу поверхню), то після досягнення потенціалу розкладу спочатку сила струму зростає лінійно, а потім практично не змінюється. Величину сили струму, яка не залежить від прикладеного потенціалу, називають граничною.
Полярографічний методом аналізу ґрунтується на залежності величини дифузійного струму від концентрації речовини у розчині, тобто на рівнянні Ільковича:
![]() або
спрощено
або
спрощено
![]() ,
,
де id – дифузійний струм, мкА; n – кількість електронів, які беруть участь в реакції; D – коефіцієнт дифузії, см2/с; m – маса ртуті, що витікає за одиницю часу, мг/с; – час утворення однієї краплі ртуті, с; C – концентрація речовини, ммоль/л.
Висота полярографічної хвилі (h) пропорційна до концентрації іонів Мn+ у розчині і її можна використати для кількісного визначення концентрації цих іонів. Значення потенціалу півхвилі Е1/2, при якому величина дифузійного струму дорівнює половині висоти хвилі, є сталим для даного іона в даних умовах полярографування. Це можна використати для якісної ідентифікації іона Мn+ у досліджуваному розчині.
Визначають концентрацію, порівнюючи висоту полярографічної хвилі досліджуваного та стандартного розчину, одержаного за таких самих умов. Найчастіше застосовують такі способи визначення концентрації: добавок, порівняння, спосіб градуйованого графіка за серією стандартних розчинів.
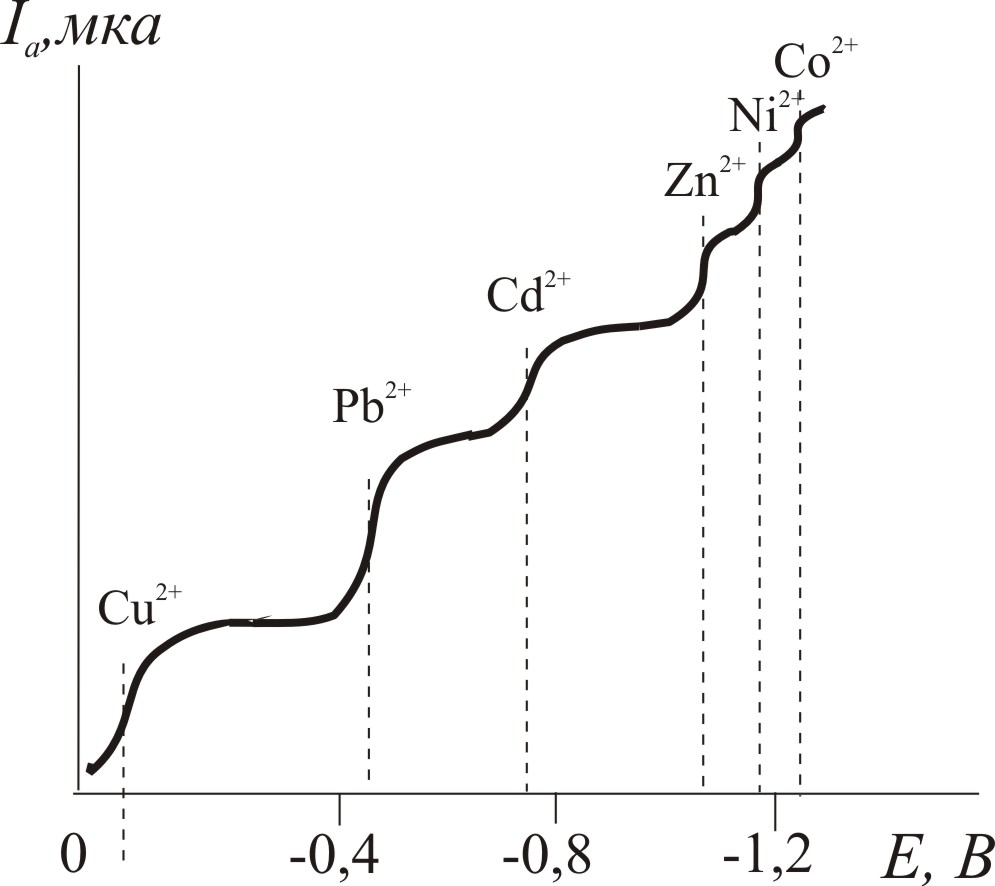
Якщо в розчині є декілька іонів, що здатні відновлюватися на ртутному крапельному електроді, то одержують криву, яка має стільки само хвиль у вигляді сходинок – полярографічний спектр (рис.10.2). Для кожної з них визначають значення Е1/2, а відтак якісний склад аналізованої суміші іонів.
Практично кожний хімічний елемент можна визначити полярографічним методом. Оптимальний інтервал концентрацій для кількісного визначення знаходиться в межах 102-104 М, що є недоліком методу. Чутливість полярографічного методу аналізу нижча, як фотометрії, відносна похибка становить 2%, а для більш розведених розчинів – 5%. Перед визначенням мікроелементів часто застосовують концентрування розчину.
|
Рис.10.2. Полярографічний спектр визначення Cu2+, Pb2+, Cd2+, Zn2+, Ni2+, Co2+ з використанням фонового електроліту KCl |
Полярографічним методом можна визначати у природних об’єктах близько 36 елементів, причому як один, так і декілька елементів з розчину. Це є перевагою методу.
Застосування полярографії для дослідження вод та витяжок
Якщо аналізують воду чи ґрунтову витяжку, то без попереднього концентрування можна визначати ті іони, яких досить багато у розчині.
У випадку аналізу витяжок, зокрема ґрунтових, попередньо хімічним обзоленням необхідно зруйнувати комплекси металів з органічними речовинами, переважно фульвокислотами.
Mn2+ визначають з розчину, але наявні у розчині солі Са понижують висоту його полярографічної хвилі відновлення.
Fe. Висоту хвилі відновлення понижує присутність солей Mg.
Cu2+, Ni2+, Zn2+, Mn2+ можна визначити без концентрування за наявності NH4Cl, якщо в ґрунтовій витяжці попередньо зв’язати іони РО43- дією ZrOCl2. Однак при цьому можливе накладання хвиль відновлення Zn2+ i Co2+. Щоб під час визначення важких металів усунути плив сполук феруму, визначення краще проводити на фоні 1М Н3РО4, яка маскує Fe3+ з утворенням [Fe(PO4)2]3-
Pb2+ визначають за наявності 3% розчину HCl;
O2 визначають за відновленням у присутності 0,15 М H2SO4 i 0,05 M (NH4)2SO4.
J- визначають окисненням до JO3- за наявності кислоти при рН 1-1,5.
Cd2+ відновлюється на ртутному крапельному електроді у присутності KNO3 при потенціалі E1/2= – 0,596 В відносно насиченого каломелевого електрода. Чутливість визначення становить 0,05 мг/л. Визначенню заважають іони Cu2+, якщо вони є у 5-кратному надлишку (їх маскують ціанідом), і не заважають іони Zn2+, Co2+, Ni2+, Mn2+, потенціал відновлення яких більш від'ємний.
Полярографічним методом можна визначати органічні речовини, які відновлюються на катоді: альдегіди, нітро- та нітрозосполуки, пероксиди. Визначення проводять часто у буферному розчині, оскільки в електрохімічному процесі беруть участь іони Н+, які відновлюються. Іншою вимогою до визначення є наявність у розчині органічного розчинника (спирту, кетону), оскільки розчинність органічних речовин у воді мала, а як фоновий електроліт використовують або сіль лужного металу, або солі тетраалкіламонію N(R)4An.
За наявності у розчині 0,1М NН4Cl визначають НСОН, СН3СОН, Н2С2О4, CO(NH2)2 при потенціалах відповідно -1,38, -1,60, -1,03, -1,55 В.
Полярографічне визначення важких металів з концентруванням.
Спочатку кислотним методом руйнують комплекси металів. Після цього важкі метали (Mo(VI), Cu2+, Zn2+, Ni2+, Co2+) розділяють екстракцією або співосадженням.
З розчину спочатку екстрагують Мо(VI) -бензоїлоксимом (або співосаджують з ним), тоді його реекстрагують розчином H2SO4, залишок обзолюють та розчиняють, в одержаному розчині визначають Мо.
Після відділення Мо(VI) з водного розчину екстрагують Cu2+, Zn2+, Ni2+, Co2+ дитизоном у CCl4. Після цього проводять реекстракцію кислотою, а тоді полярографічно визначають метали. Для переведення Со2+ з дитизонату у водний розчин проводять повторну реекстракцію кислотою.
У розчині після реекстракції визначають Cu2+ у присутності CH3COONa при потенціалі -0,3 В. До розчину додають KSCN і визначають інші іони: Ni2+ (при потенціалі -0,72 В), Zn2+ (при -1,09 В), Co2+ (при -1,45В).
Складність полярографічного визначення кобальту полягає в дуже низькому потенціалі відновлення Co2+ та дуже малій концентрації його в природних об’єктах. Тому після екстракційного концентрування важких металів та реекстракції полярографічно визначають Cu2+, Zn2+, Ni2+. Після повторної реекстракції визначають Со2+фотометрично з нітрозо-R-сіллю.
Сучасні варіанти полярографії дозволяють визначати до 1103 мкг/мл. Аналіз можна провести з об’єму 12 мл, а інколи навіть з однієї краплі розчину.
Амперометричним титруванням визначають у стічних водах низку аніонів: ClO-, CN-, SCN-, Cl-, Br-, SO42-, SO32-, S2-. При цьому не обов’язково видаляти з розчину кисень та вводити поверхнево-активні речовини.
SO42- титрують у нейтральному середовищі розчином солі Pb2+, який відновлюється на катоді. Визначенню заважає присутність у розчині хромату та дихромату.
Cl-, Br-, J-, SCN- титрують на Pt електроді розчином Hg2(NO3)2 або AgNO3, з якого на електроді відновлюється катіон металу. Титрування проводять за наявності 0,1M KNO3, ізоамілового та етилового спирту (15:1) при рН 2-3;
AsO32-, який окиснюється на аноді, титрують розчином BrO3-.
За іншим способом AsO32- титрують у нейтральному середовищі розчином J2, а на катоді йод відновлюється до J-.
AsO43- у розчині титрують в кислому середовищі розчином J-, а виділений при цьому J2 відновлюється на катоді.
Ag+ в кислому середовищі титрують розчином J-, який окиснюється на аноді.
Полярографія каталітичних струмів
Фактором, який визначає величину дифузійного струму, є швидкість хімічної реакції, що протікає біля електроду. Відбувається хімічна взаємодія, у якій речовина або каталізатор є електрохімічно активним:
1)
А![]() В, В+Х
В, В+Х
![]() А.
А.
2) А+Х АХ, АХ А+Х, де Х – каталізатор.
Хімічна реакція характеризується константою рівноваги K. Відновлені на електроді іони каталізатора регенеруються і знову надходять у хімічну реакцію, тому сила дифузійного каталітичного струму (іК) в 10-100 разів більша за силу дифузійного струму, і, отже, чутливість підвищується. Сила дифузійного каталітичного струму залежить від концентрації речовини СВ:
![]() ,
,
де n – число електронів, які беруть участь в реакції; k – константа швидкості хімічної реакції; SHg – середня площа поверхні ртутного електрода, – товщина шару, в якому відбувається регенерація; SHg – об’єм реакційного шару; СоВ, СоХ – початкова концентрація речовини та каталізатора.
Кількісне визначення залежить від константи швидкості реакції, а остання від температури, тому необхідно комірку термостатувати.
Каталітичні струми відновлення відомі для багатьох органічних речовин та неорганічних іонів: феруму, плюмбуму, кобальту, титану, урану, вольфраму, молібдену.
Визначення молібдену у воді та ґрунтовій витяжці. Для визначення вмісту рухливих форм молібдену одержують ґрунтову витяжку дією розчину оксалатної кислоти, випаровують її, а залишок прожарюють при 450С щонайменше 4 год для повного розкладу органічних речовин. Після цього залишок розчиняють у концентрованій HCl. Іони V(V) i Cr (VI), які заважають визначенню, відновлюють дією Fe2+. Після цього Мо (VI) екстрагують -бензоїлоксимом у хлороформі. Хлороформ відганяють, екстракт поміщають у тигель і прожарюють при 450С до повного руйнування органічних речовин. Визначення молібдену проводять різними способами.
За першим способом до одержаного охолодженого залишку доливають фоновий електроліт – кислоти-окисники, і полярографують. Каталітичний процес складний, cпрощено його можна представити такими реакціями за участю окисника NO3-:
Н2МоО4*
+ 6Н++
4![]() Mo2+
+ 4Н2О,
Mo2+
+ 4Н2О,
3Mo2++4NO3- + 4H2О 3Н2МоО4 +4NO+2H+.
Якщо як фон використати 1M KNO3+1MH2SO4 або 1,6 М HClO3, можна виявити молібден у концентрації 0,05 мг/л, якщо 2,4 M HNO3, то 0,005 мг/л.
За другим способом, після аналогічної підготовки проби в розчин молібдату додають 0,05 M H2O2 і 1М фосфатний буферний розчин при рН 4. Утворюється комплекс молібдату з пероксидом MoO2(H2O2)HPO4, з якого на катоді відновлюється пероксид. Процеси у розчині та на електроді можна представити такими рівняннями:
H2MoO4+H3PO4MoO2HPO4+2H2O та MoO2HPO4+H2O2MoO2(H2O2)HPO4*,
MoO2(H2O2)HPO4*+2H++2 MoO2HPO4+2H2O.
Полярографування проводять при потенціалі +0,1 – +0,3 В, причому кисень з розчину не видаляють. Чутливість визначення – 0,0005 мг/л.
Інверсійна вольтамперометрія
Цей різновид вольтамперометрії використовують для визначення дуже малих кількостей речовин у розчині. Метод ґрунтується на електролітичному виділенні речовини з розчину, а потім, при зміні потенціалу на протилежний, на розчиненні речовини з поверхні електрода і переходу знову в розчин. Під час електролізу при E>E(граничного) на електроді відбувається:
відновлення
Pb2+
+ 2![]()
Pb, або
Pb, або
окиснення Mn2+ + 2H2O – 2 MnO2 + 4H+,
при якому речовина накопичується на ньому.
При плавній зміні потенціалу у зворотному напрямку, відбувається протилежний процес (окиснення або відновлення), при якому речовина розчиняється з електрода. Отже, у процесі інверсійного вольтамперометричного визначення відбувається концентрування речовин. Таким чином інверсійна вольтамперометрія є одним з найбільш чутливих методів. В аналізі використовують такі основні процеси.
Відновленням на електроді катіонів з наступним анодним розчиненням визначають більшість важких металів. Умови визначення: переведення іонів з неелектроактивних у активні форми (руйнування комплексів); підвищення чутливості із застосуванням концентрування розчину. Спочатку Sn2+ співосаджують на Mg(OH)2, а Cd2+, Pb2+, Cu2+ концентрують екстракцією дитизонатів.
Відновлення (окиснення) іонів зі змінним ступенем окиснення з утворенням малорозчинної сполуки.
Наприклад, J- на електроді окиснюється до йоду (2J-–2 J2), який адсорбується барвником трифенілметанового ряду з утворення малорозчин-ного J2ТФМБ. Під час наступного відновленні йоду комплекс руйнується. Якщо у розчині був J2, то його попередньо відновлюють гідразин-гідрохлоридом до J-, а потім визначають. Чутливість – 410-9 моль/л.
Відновлення (окиснення) активного електрода, при якому утворюється (розчиняється) осад солей або гідрооксидів.
Відновлення (окиснення) твердих розчинів, механічно введених в об’єм електрода (амальгами).
Метод, при якому спочатку відбувається електровиділення як результат відновлення, а потім визначення в результаті окиснення, називають анодною інверсійною вольтамперометрією, а якщо процеси проходять протилежно, то це катодна інверсійна вольтамперометрія.
Для визначення важких металів найчастіше використовують процес катодного відновлення. Під час електролізу протягом певного часу катіони металів з розчину виділяються на катоді. Після цього їх окиснюють з електрода і визначають. Переважно метали визначають на ртутному електроді, при цьому утворюється амальгама з металами.
Приклавши до ртутного катода значення потенціалу, при якому відновлюється катіон з розчину, при перемішуванні розчину проводять електроліз. Катіон металу відновлюється на катоді:
Men++n +Hg Hg(Me),
сила
струму відновлення:
![]() ,
,
де Со – початкова концентрація іона металу в розчині; n – кількість електронів, які беруть участь в окисно-відновному процесі; Кел. – константа процесу електровідновлення металу.
Після відновлення катіона і одержання амальгами поступово змінюють напругу в протилежному напрямі. Починається процес окиснення металу з амальгами, що відповідає силі струму анодного окиснення (iaн):
Hg(Me)– ne = Hg +Men+.
Одержують криву анодного розчинення (рис.10.3).
|
Рис.10.3. Крива анодного розчинення металу з амальгами. |
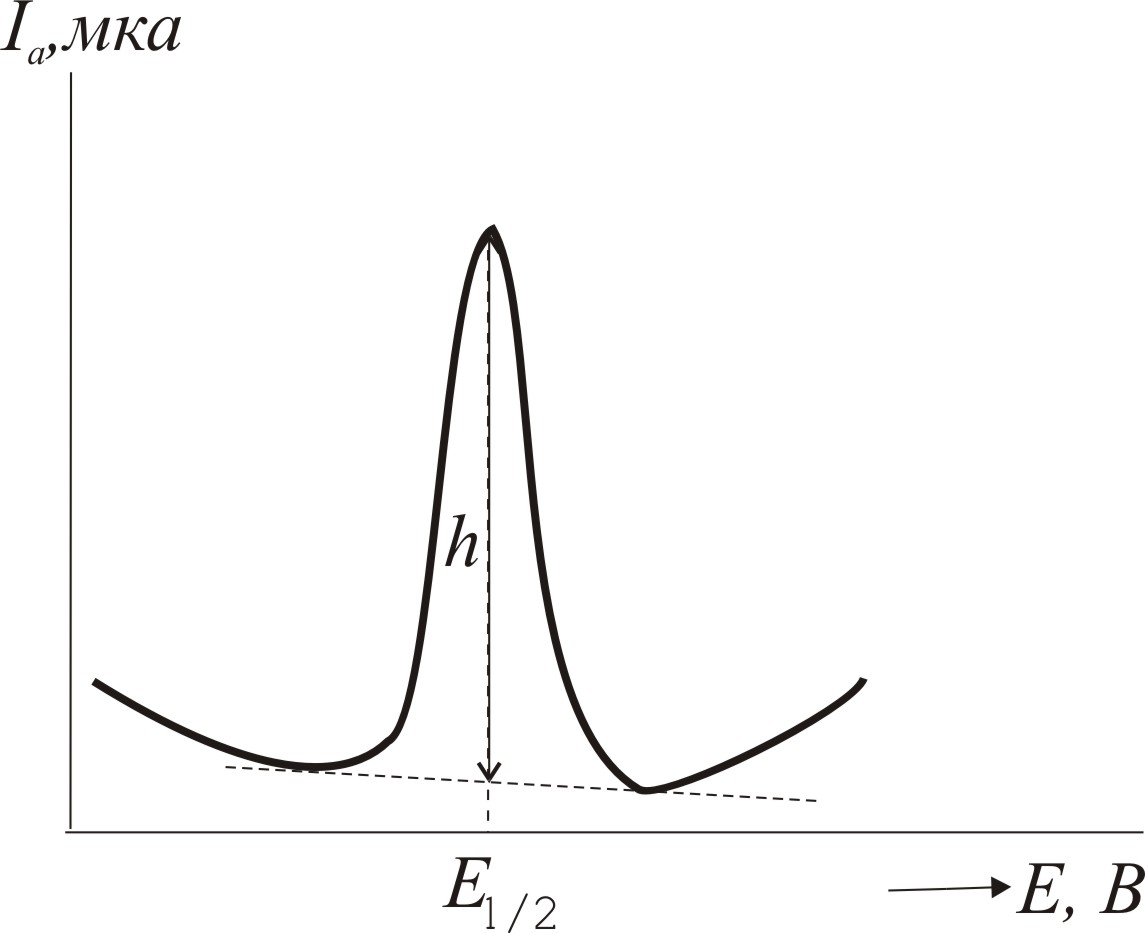
Якісне виявлення проводять за величиною потенціалу піку кривої, а кількісне – за його висотою h.
Величина анодного струму залежить від умов проходження електролізу: об’єму розчину; величини ртутної краплі; тривалості електролізу, усі ці параметри об’єднуються поправкою (b).
Якщо об’єм розчину великий, а ртутна крапля і час електролізу малі, тоді СоCкінцеве, (b<0,1) і силу струму анодного окиснення обчислюють так:
 .
.
Якщо об’єм розчину малий, ртутна крапля та час електролізу великі, тоді Со>Cкінцеве (b>3), силу струму анодного окиснення обчислюють:
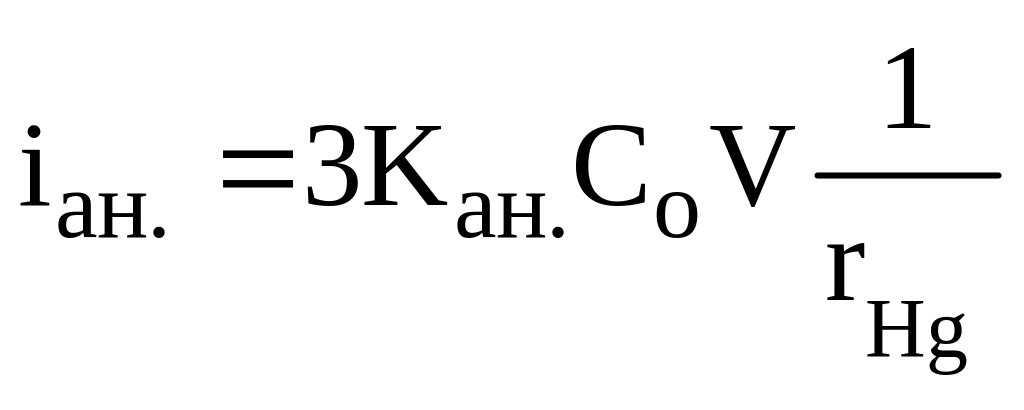 .
.
Для проміжних умов (3>b>0,1) сила анодного струму:
![]() ,
,
де Со – початкова концентрація іона металу в розчині; V – об’єм розчину, мл;
r(Hg) – розмір краплі ртуті; n – кількість електронів, які беруть участь в окисно-відновному процесі; F – стала Фарадея; – тривалість електролізу, с;
Кан. – константа процесу електроокиснення металу.
Концентрацію в методі інверсійної вольтамперометрії визна-чають за порівнянням висот піків окиснення (відновлення) стан-дартного та досліджуваного розчину, за методом добавки.
Для визначення різних елементів застосовують різні електроди. Зокрема As, Bi, Sb визначають на графітовому електроді. Графітовий електрод дає відтворювані результати при роботі впродовж 1-1,5 год, а потім, у присутності поверхнево-активних речовин, цей час сильно зменшується до 30-40 хв. Важкі метали визначають на ртутному електроді: Pb2+ на стаціонарній краплі Hg; Cd2+, Cu2+, Zn2+ – на крапельному електроді; Ag+ – на амальгамованому електроді; використовують як електрод завислу краплю Hg на Ag дротині чи на контакті з Ag, Au, Pt.
Особливості аналізу стічних вод та природних об’єктів
При аналізі природних вод можна не використовують фону, оскільки солі макроелементів створюють належний фон електролітів.
Якщо аналізують ґрунтові витяжки, то спочатку кислотою руйнують органічні речовини, а тоді залишок розчиняють.
Одночасне визначення декількох металів методом інверсійної вольтамперометрії можна провести з одного розчину, якщо електроліз проходить при такому від’ємному потенціалі, який відповідає найважче відновлюваному катіону. Коли після завершення електровиділення всіх металів плавно підвищують потенціал, то анодне окиснення відбувається послідовно. Потенціал при досягненні кожного піку окиснення не підвищують доти, поки повністю не розчиниться метал. При правильно підібраному фоні досягають селективного визначення всіх металів, а за висотами піків окиснення визначають їхню кількість.
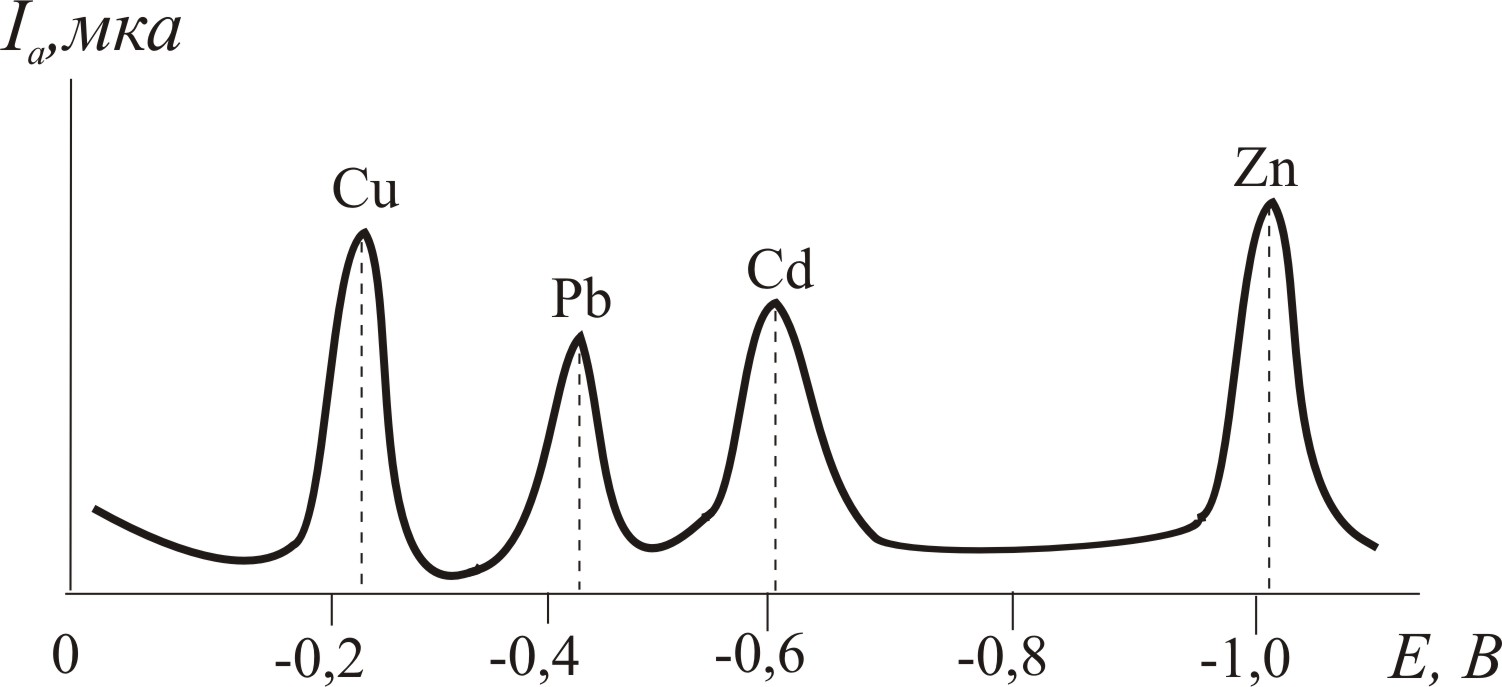
Прикладом може бути визначення у стічній воді солей Cu2+, Zn2+, Cd2+, Pb2+ (рис. 10.4). У розчині природного об’єкта спочатку відновлюють Fe3+ до Fe2+ аскорбіновою кислотою. Тоді у присутності HCl визначають Cu2+, Cd2+, Pb2+. Розчин підлужнюють до рН 4–5 і в цих умовах визначають Zn2+.
Якщо як фоновий електроліт використовувати HCl+уротропін при рН 5, то безпосередньо з розчину визначають всі іони: Cu2+, Cd2+, Pb2+, Zn2+. Іноді перед визначенням ці важкі метали концентрують екстракцією і визначають після реекстракції. Чутливість визначень: Cu2+– 210-8, Pb2+ – 610-9, Cd2+ – 510-10 моль/л.
|
Рис.10.4. Анодна інверсійна вольтамперограма стічної води (електроліз при -1,2В на ртутно-графітовому електроді) |
На Hg електроді методом катодної інверсійної вольтамперометрії визначають також аніони, які утворюють важкорозчинні сполуки з іонами Hg2+, що утворюються при анодному окисненні ртуті. Зокрема можна визначити такі аніони: J-, Cl-, Br-, S2-, SCN-.