

1.6.3. Синтез кетонов из нитрилов карбоновых кислот реакцией Гриньяра
В нитрилах карбоновых кислот атом углерода цианогруппы является электрофильным центром, к которому могут присоединяться нуклеофильные реактивы Гриньяра. Продукт этого присоединения при гидролизе превращается в так называемый имин, который далее гидролизуется до кетона.

Так, например, ацетофенон (метилфенилкетон) можно получить взаимодействием ацетонитрила (нитрила уксусной кислоты) и фенилмагнийбромида с последующим гидролизом.
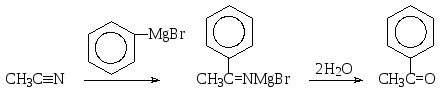
Возможен и другой вариант синтеза ацетофенона реакцией Гриньяра: из бензонитрила (нитрила бензойной кислоты) и метилмагнийиодида.
2. Химические свойства
Химическое поведение альдегидов и кетонов обусловлено наличием очень полярной карбонильной группы (дипольный момент связи С=О около 2,5 D).
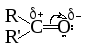
Относительно большой частичный положительный заряд на атоме углерода придает альдегидам и кетонам электрофильные свойства, поэтому основной тип реакций этого класса соединений – нуклеофильное присоединение (AdN) по карбонильной группе.
2.1. Кислотно-основные свойства и кето-енольная таутомерия
Альдегиды и кетоны, имеющие в α-положении к карбонильной группе хотя бы один атом водорода, проявляют заметные кислотные свойства (рКа ~20), поскольку сопряженное основание стабилизировано р-π-сопряжением.
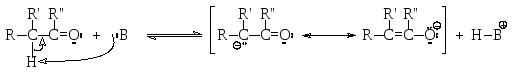
граничные структуры
сопряженного основания
Для таких альдегидов и кетонов возможна кето-енольная таутомерия. Кето-енольная таутомерия – это явление, связанное с существованием в динамическом равновесии двух (или более) структурных изомеров, отличающихся расположением атома водорода – или у атома углерода в α-положении, или у атома кислорода – и распределением π-электронной плотности – или между атомами углерода и кислорода карбонильной группы, или между карбонильным атомом углерода и α-атомом углерода. Это явление легко понять, если представить себе процесс протонирования сопряженного основания: а именно, протон может присоединиться как к атому углерода, от которого он был оторван основанием В, так и к атому кислорода, на котором во второй граничной структуре сопряженного основания локализован отрицательный заряд.
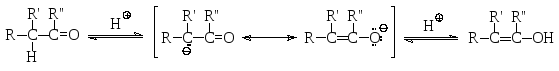
альдегид или кетон енол
Положение кето-енольного равновесия зависит от строения карбонильного соединения. Для обычных альдегидов и кетонов оно сильно сдвинуто в сторону значительно более термодинамически устойчивой карбонильной формы. Так, в обычных условиях в кето-енольном равновесии для такого кетона, как ацетон, содержится всего лишь 2,4∙10-4% енола.
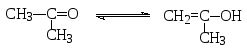
При возможности стабилизации енольной формы ее содержание в равновесии может быть и значительно больше. Ацетоуксусный эфир (этиловый эфир 3-оксобутановой кислоты) в обычных условиях представляет собой смесь кетонной и енольной форм, в которой представлено около 7% последней. По сравнению с ацетоном это почти в 30000 раз больше, что объясняется термодинамической стабилизацией енольной формы в результате образования обширной системы р-π-π-сопряжения и внутримолекулярной водородной связи.

Если в α-положении к карбонильной группе атомы водорода отсутствуют, то такой альдегид или кетон заметных кислотных свойств не проявляет, и, разумеется, кето-енольная таутомерия для такого альдегида или кетона невозможна. Например, формальдегид или бензальдегид не имеют в α-положении к карбонильной группе атомов водорода (в формальдегиде вообще нет α-положения), поэтому кислотность этих соединений чрезвычайно мала, и кето-енольной таутомерии для них нет.
Основность альдегидов и кетонов обусловлена относительной доступностью неподеленной пары электронов атома кислорода карбонильной группы.
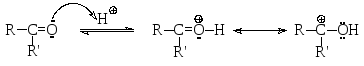
Основность альдегидов и кетонов невысока, однако она играет заметную роль в реакциях нуклеофильного присоединения, поскольку в протонированной форме электрофильность атома углерода значительно выше. Поэтому реакции нуклеофильного присоединения могут катализироваться кислотами.