

4. АВТОМАТИЧЕСКИЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ (ОФС 42-0076-08)
Метод автоматического элементного анализа основан на высокотемпе-
ратурном (от 1100 до 1800 ºС) окислительном разложении исследуемых ве-
ществ с последующим хроматографическим определением компонентов об-
разовавшейся газовой смеси.
Метод элементного анализа может быть использован для определения содержания активного вещества в фармацевтических субстанциях, в состав молекул которых входят углерод (С), водород (Н), азот (N) или сера (S), на основании данных элементного анализа на любой из этих элементов. Приме-
нение метода элементного анализа для определения других элементов дол ж-
но быть описано в частной фармакопейной статье. Метод может быть ис-
пользован и для целей идентификации лекарственного вещества на основе установления его брутто-формулы.
Для определения содержания элементов C, H, N и S в субстанциях про-
водят высокотемпературное окислительное разложение в потоке гелия, либо его смеси с кислородом в присутствии катализатора окисления, последующее восстановление окислов азота до молекулярного азота в присутствии катали-
затора восстановления и определение образующихся продуктов (CO2, H2O, N2, SO2), соответствующих определяемым элементам, методом газовой хро-
матографии.
Метод элементного анализа может быть применен и для определения содержания активного вещества в лекарственных препаратах, но только на основании определения азота, входящего в состав молекулы фармацевтиче-
ской субстанции, при условии отсутствия азота в молекулах вспомогатель-
ных веществ. Вследствие присутствия наполнителей при окислительном раз-
ложении лекарственных препаратов образуется большое количество углерода диоксида (СО2), мешающего определению азота. В связи с этим газохромато-
графическое определение азота проводится после предварительного погло-
щения углерода диоксида (вместе с серы диоксидом) и воды.
2
В качестве катализатора окисления обычно используют меди(II) оксид
(CuO) с добавкой ванадия(V) оксида (V2O5) или посеребрянного кобальта(II,III) оксида (Co3O4). В качестве катализатора восстановления при-
меняют электролитическую медь. Применение других катализаторов должно быть указано в частной фармакопейной статье.
Для поглощения диоксидов углерода и серы (CO2 и SO2) используют ловушки с натронной известью, воды – с ангидроном или освобождаются от воды в соединительных трубках, стенки которых селективно проницаемы для воды.
Метод применим для анализа как твердых, так и жидких лекарствен-
ных средств.
Прибор. Определение проводят на приборе «автоматический элемент-
ный анализатор», основными составными частями которого являются: ульт-
рамикровесы, узел ввода пробы – автодозатор капсулированных (запечатан-
ных в контейнеры из оловянной фольги) проб анализируемых образцов,
окислительный и восстановительный реакторы, помещенные в электропечь,
ловушки (поглотители), хроматографическая колонка, детектор по теплопро-
водности и система для обработки данных и управления прибором.
Методика. При анализе твердых лекарственных средств их предвари-
тельно тщательно растирают в агатовой ступке.
В качестве стандартных образцов используют ацетанилид, цистеин, ме-
тионин с установленным содержанием элементов – стандартные образцы для микроанализа.
Точные навески (от 0,5 до 1,5 мг стандартного образца или субстанции или от 1 до 5 мг препарата), взятые на ультрамикровесах с точностью до
0,001 мг, помещают в предварительно взвешенные пустые оловянные кон-
тейнеры. Запечатывают контейнеры с помощью специального устройства,
взвешивают капсулированные образцы и помещают в кассету автодозатора.
При увеличении объемов катализаторов окисления и восстановления навеска

3
может быть увеличена в 5–10 раз, при этом точность взвешивания может со-
ставлять 0,01 мг.
Проводят контрольный опыт, для чего с помощью автодозатора в реак-
тор вбрасывают пустые оловянные контейнеры (число проб не больше трех);
при этом регистрируется содержание определяемого элемента для каждой из них. Затем последовательно сжигают по 3–4 навески капсулированных об-
разцов (стандартного и испытуемого).
По полученным значениям площадей пиков стандартных образцов с учетом значения контрольного опыта автоматически строится градуировоч-
ный график и рассчитывается поправочный коэффициент K к площади пика
определяемого элемента по формуле:
|
|
K = |
y ао |
|
|
|
|
, |
|
|
|
(Sо–Sк) 100 |
||
где: y |
– |
содержание определяемого элемента в стандартном образце, в |
||
|
|
процентах; |
|
|
Sо |
– |
площадь пика на хроматограмме стандартного образца; |
||
Sк |
– |
площадь пика на хроматограмме контрольного опыта; |
||
ао |
– |
навеска стандартного образца в миллиграммах. |
||
Содержание определяемого элемента в испытуемом образце лекарс т-
венного средства (субстанции, препарате) в процентах (Хэ) автоматически
рассчитывается по формуле: |
|
|
|
Хэ = |
(S –Sк) × K × 100 |
, |
|
a |
|||
|
|
где: S – площадь пика на хроматограмме испытуемого образца;
а– навеска порошка лекарственного средства (для субстанции в пересчете на сухое или безводное вещество), в миллиграммах.
Содержание лекарственного вещества в субстанции в процентах (Х)
вычисляют по формуле:
Х = Хэ× М.м. , A.м. × n
где: М.м. – молекулярная масса лекарственного вещества;
4
А.м. – атомная масса определяемого элемента;
n– число атомов определяемого элемента в молекуле лекарственного вещества.
Содержание лекарственного вещества в одной дозе препарата в милли-
граммах (Х) вычисляют по формуле:
Х = |
Хэ × М.м. × G |
, |
|
100 × A.м. × n |
|||
|
|
где: G – средняя масса одной дозы препарата в миллиграммах.
Примечания
1.Навеску анализируемого лекарственного средства выбирают такой, чтобы количество определяемого элемента, образовавшееся от сжигания навески испытуемого образца, было близко к количеству, образующемуся при сжигании навески стандартного образца.
2.Используемые реактивы
Ацетанилид, цистеин, метионин – стандартные образцы для элементного анализа.
Меди(II) оксид (CuO). (М.м. 79,545). Кусочки проволоки длиной 1–3 мм, толщиной около, 1 мм серого цвета. Поставляется фирмойпроизводителем элементного анализатора.
Кобальта(II,III) оксид (Co3O4) посеребрянный. Гранулы черного цвета диаметром около 1 мм. Поставляется фирмой-производителем элементного анализатора.
Ангидрон. Mg(ClO4)2. (М.м. 223,20). Магния перхлорат.
Белая пористая масса, очень энергично поглощающая влагу (до 60 % от своей массы) с образованием кристаллогидрата Mg(ClO4)2·6Н2О; растворяется в воде с выделением теплоты, при поглощении воды не расплывается. Применяется для глубокой осушки газов от воды. Может быть обезвожен нагреванием в вакууме до 200–250 °С и повторно использован.
Натронная известь. Смесь натрия гидроксида NaOH и гашеной извести Са(ОН)2. Натровая известь. Натристая известь.
Белая пористая масса. Поглощает воду (влагу) и углерода диоксид из воздуха, переходя в смесь карбонатов натрия и кальция.
5. ОПРЕДЕЛЕНИЕ РАСПРЕДЕЛЕНИЯ ЧАСТИЦ ПО РАЗМЕРУ МЕТОДОМ ЛАЗЕРНОЙ ДИФРАКЦИИ СВЕТА (ОФС 42-0083-08)
Метод лазерной дифракции света, используемый для определения рас-
пределения частиц по размеру, основан на анализе профиля рассеяния света,
возникающего при освещении частицы коллимированным лазерным пучком.
Традиционно метод позволяет измерять частицы в диапазоне от 0,1 мкм до 3 мм. Современные достижения в приборостроении позволили расширить этот диапазон (от 0,1 мкм до 8 мм).
Метод предназначен для контроля качества лекарственных препаратов
(порошки, суспензии, эмульсии, пасты, настойки и т. д.) по показателю «раз-
мер частиц и их распределение». Специфические условия проведения анали-
за по измерению размера частиц и их распределению в конкретных лекарст-
венных средствах указывают в соответствующих частных фармакопейных статьях. Метод позволяет определять, а затем и нормировать размер частиц и их распределение в субстанциях.
Метод не может отличить рассеяние от отдельных частиц и рассеяние от кластеров частиц, т. е. агломератов или агрегатов. В случае если образцы содержат агломераты или агрегаты частиц и если необходимо определить распределение отдельных частиц по размеру, то перед измерением кластеры диспергируют на отдельные частицы. Для несферических частиц получают соответствующее распределение эквивалентных сфер по размеру, поскольку метод предполагает использование сферических частиц в своей оптической модели. Полученное распределение частиц по размеру может отличаться от распределений, основанных на других физических принципах (например, се-
диментации или ситовом определении).
Принцип. Образец, диспергированный в жидкости или газе с необхо-
димой концентрацией, помещается в лазерный пучок. Свет, рассеянный от частиц на различных углах, измеряется многоэлементным детектором. Чис-
ленные значения, представляющие профиль рассеяния света, регистрируются
для последующего анализа. В дальнейшем эти значения математически пре-
образуются с помощью оптической модели в доли от общего объема отдель-
ных размерных классов, формируя, таким образом, объемное распределение частиц по размеру.
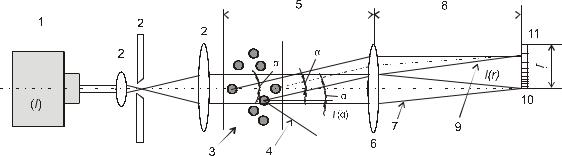
Схема прибора для лазерной дифракции представлена на рис. 5.1.
Взаимодействие луча падающего света и частиц дисперсной фазы при-
водит к образованию профиля рассеяния света с разными значениями интен-
сивности света при различных углах. Общее распределение угловой интен-
сивности, состоящее из прямого и рассеянного света, фокусируется линзой на многоэлементном детекторе. Линза создает профиль рассеяния света, ко-
торый не зависит от расположения частиц в световом луче.
Рис. 5.1. Схема прибора определения размера частиц методом лазерной д и- фракции
1 – источник лазерного излучения, 2 – модуль обработки лазерного излучения, 3 – частицы, 4 – рассеянный свет, не собранный линзой (6), 5 – рабочее расстояние линзы (6),6 – линза Фурье, 7 – прямой луч, 8 – фокусное расстояние линзы (6), 9 – рассеянный луч, 10 – детектор затемнения, 11 – многоэлементный детектор
Пробоподготовка. Методика пробоподготовки должна обеспечивать получение репрезентативного образца требуемого объема для измерения размера частиц.
Спреи, аэрозоли и пузырьки газа в жидкости измеряются непосредст-
венно, поскольку пробоподготовка или разведение могут изменить распреде-
ление частиц по размеру.
Сыпучие порошки также можно преобразовать в аэрозоли при помощи диспергаторов, использующих энергию сжатого газа или давление вакуума.
Полученный аэрозоль проходит через зону измерения, после чего попадает во впускное отверстие вакуумного блока, где частицы аэрозоля собираются.
В качестве дисперсионной среды могут быть использованы вода и раз-
личные органические растворители (этиловый спирт, метиловый спирт, изо-
пропиловый спирт, гексан, ацетон, толуол и другие), что должно быть отраже-
но в частной фармакопейной статье.
Определение диапазона концентрации. Для того чтобы получить приемлемое соотношение сигнал-шум в детекторе, концентрация частиц в дисперсии должна превышать минимальный уровень. Также, она должна быть меньше максимального уровня для избежания многократного рассеяния.
На диапазон концентрации влияют: ширина лазерного луча, расстоя-
ние, проходимое лучом лазера в зоне измерения, оптические свойства частиц и чувствительность элементов детектора. Измерения необходимо проводить при различных концентрациях частиц для определения оптимального диапа-
зона концентрации для каждого характерного образца материала.
Методика определения. Измерение размеров частиц осуществляют на малоугловых измерителях дисперсности (например, на приборе типа МИД-5)
в соответствии с руководством по эксплуатации прибора и инструкцией поль-
зователя.
После соответствующей регулировки оптической части прибора, долж-
но быть произведено фоновое измерение среды, в которой отсутствуют дис-
персные частицы. Уровень сигнала фона должен быть ниже соответствующе-
го порогового значения. После проведения фонового измерения проводят измерение пробы. Обычно при измерении проводится большое число регист-
раций сигнала на элементах детектора и определяется среднее значение для каждого элемента. Положение и размер элементов детектора, фокусное рас-
стояние линзы определяют диапазон углов рассеяния для каждого элемента.
Большинство приборов также измеряют интенсивность центрального луча. Различие интенсивностей центрального луча в дисперсной системе и
фонового измерения является параметром затемнения и свидетельствует о величине рассеянного света и концентрации частиц. Результаты измерения сохраняются в памяти компьютера.
Выбор оптической модели. В большинстве случаев применяются ап-
проксимация Фраунгофера или теория Ми. При размере частиц менее
25 мкм различия между оптическими моделями становятся более существен-
ными. В этом диапазоне более точные результаты позволяет получать теория Ми. При использовании теории Ми в прибор необходимо ввести значения показателя преломления частиц и среды или их отношение.
Повторные измерения. Число повторных измерений зависит от кон-
кретного материала. Обычно измеряют не менее трех репрезентативных об-
разцов одной серии.
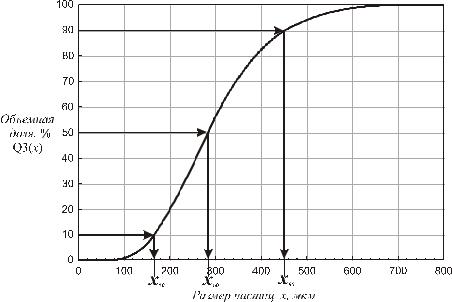
Результаты измерений. Результаты обычно представляют в виде инте-
грального объемного распределения частиц по размеру (рис. 5.2). Величины xm
отражают размер частиц, где m – это доля частиц с размером x и менее. Для оценки распределения по размеру обычно используют значения x10, x50 и x90.
Рис. 5.2. Интегральное объемное распределение частиц по размеру x – размер частиц, определяемый как диаметр объема эквивалентной сферы; Q3(x) – объемная доля частиц с размером x и менее; x10, x50, x90 – размер частиц, соответствующий объемной доле 10, 50 и 90 % соответственно
Калибровка прибора. Работа прибора базируется на основных прин-
ципах рассеяния лазерного излучения, при условии идеализированных свойств частиц, поэтому калибровка прибора перед измерением не требуется.
Проверку правильности работы прибора можно осуществить путем измерения сертифицированного стандартного материала, состоящего из частиц известно-
го распределения по размеру.
Поверка системы. Работу прибора необходимо подтверждать через регулярные интервалы времени или с надлежащей частотой. Поверку систе-
мы производят с использованием контрольного материала, известного рас-
пределения по размеру. Средние значения трех измерений должны отличаться от установленного значения не более чем на 10 % для x50 и не более чем на 15 %
для x10 и x90. Для x < 10 мкм, эти величины необходимо удвоить.
Повторяемость. Предпочтительно использование сертифицированных или стандартных материалов, состоящих из сферических частиц известного распределения по размеру, с размерными группами, отличающимися по разме-
ру более чем в 10 раз. Действие прибора считается соответствующим требова-
ниям, если среднее значение x50 по крайней мере трех независимых измере-
ний сертифицированного или стандартного материала отличается не более чем на 3 % от установленного диапазона значений. Средние значения x10 и x90
не должны превышать установленный диапазон значений более чем на 5 %. Коэффициент вариации должен быть менее 3 % для x50 и менее 5 % для x10 и x90. Для x < 10 мкм, эти величины необходимо удвоить.
Воспроизводимость. Воспроизводимость метода главным образом зависит от характеристик материала (измельченный/неизмельченный, твер-
дый/ломкий), а также от лекарственной формы. Обычно измеряют не менее трех репрезентативных образцов одной серии. Коэффициент вариации дол-
жен быть менее 10 % для x50. Для значений x10 и x90 коэффициент вариации должен быть менее 15 %. Для x < 10 мкм, эти величины необходимо удвоить.
Меры предосторожности. При проведении измерений жидких дис-
персий необходимо избегать появления пузырьков воздуха, испарения жид-
кости или других неоднородностей дисперсии. При работе с сухими диспер-
сиями избегать неравномерного потока частиц от диспергатора или турбу-
лентного воздушного течения. Такие эффекты могут вызывать ложные рас-
пределения частиц по размеру.