

Micro-Nano Technology for Genomics and Proteomics BioMEMs - Ozkan
.pdf406 |
PHILIP SANTANGELO ET AL. |
signal-to-background ratio, coupled with an improved specificity, has allowed molecular beacons to enjoy a wide range of biological and biomedical applications.
A conventional molecular beacon has four essential components: loop, stem, fluorophore, and quencher, as illustrated in Figure 12.1a. The loop usually consists of 15–25 nucleotides and is selected based on target sequence and melting temperature. The stem, formed by two complementary short-arm sequences, is typically 4–6 bases long and is usually chosen to be independent of the target sequence (Figure 12.1B). Molecular beacons, however, can also be designed such that one arm of the stem participates in both stem formation and target hybridization (shared-stem molecular beacons), as illustrated schematically in Figure 12.1C. Although a molecular beacon can be labeled with any desired reporter-quencher pair, proper selection of the reporter and quencher could improve the signal-to-background ratio and multiplexing capabilities.
There are two major design issues of nanostructured molecular probes for gene detection: specificity and melting temperature. First, to ensure specificity, for each target gene, one can use NCBI BLAST [17] or similar software to select 16–20 base target sequences that are unique for the target mRNA. Secondly, since the melting temperature of molecular beacons affects both the signal-to-background ratio and detection specificity, especially for mutation detection, one has to systematically adjust the G-C content of the target sequence, the loop and stem lengths and the stem sequence of the molecular beacon to realize the optimal melting temperature. In particular, it is necessary to understand the effect of molecular beacon design on melting temperature so that, at 37◦C, single-base mismatches in target mRNAs can be differentiated. In the case of homogeneous assays, secondary structure of mRNA is not an issue due to the ability to denature the mRNA structure via temperature.
The loop, stem lengths and sequences are the critical design parameters for molecular beacons, since at any given temperature they largely control the fraction of molecular beacons in each of three different conformational states: bound-to-target, stem-loop, and random-coil [16]. In many applications, the choices of the probe sequence are limited by target-specific considerations, such as the sequence surrounding a single nucleotide polymorphism (SNP) of interest. However, the probe and stem lengths, and stem sequence, can be adjusted to optimize the performance (i.e., specificity, hybridization rate and signal- to-background ratio) of a molecular beacon for a specific application [15, 18].
In general, it has been found that molecular beacons with longer stem lengths have an improved ability to discriminate between wild-type and mutant targets in solution over a broader range of temperatures. This can be attributed to the enhanced stability of the molecular beacon stem-loop structure and the resulting smaller free energy difference between closed (unbound) molecular beacons and molecular beacon-target duplexes, which generates a condition where a single-base mismatch reduces the energetic preference of probe-target binding. The competition between the two stable conformations of a molecular beacon (i.e., closed and bound to target) also explains why it has an enhanced specificity compared with linear probes. Longer stem lengths, however, are accompanied by a decreased probe-target hybridization kinetic rate. Similarly, molecular beacons with short stems have faster hybridization kinetics but suffer from lower signal-to-background ratios compared with molecular beacons with longer stems. It is interesting to note, however, that stem-less molecular beacons, which lack the short complementary arms and rely solely on the random-coiled nature and interactions between the dye and quencher to maintain a dark state, are still able to differentiate between bound and unbound states [19].
408 |
PHILIP SANTANGELO ET AL. |
Selecting a fluorophore label for a molecular beacon as the reporter is usually not as critical as the hairpin probe design since many conventional dyes can yield satisfactory results. However, proper selection could yield additional benefits such as an improved signal- to-background ratio and multiplexing capabilities. Since each molecular beacon utilizes only one fluorophore it is possible to use multiple molecular beacons in the same assay, assuming that the fluorophores are chosen with minimal emission overlap [20]. Molecular beacons can even be labeled simultaneously with two fluorophores, i.e., “wavelength shifting” reporter dyes, allowing multiple reporter dye sets to be excited by the same monochromatic light source yet fluorescing in a variety of colors [21]. Specifically, a “harvester” fluorophore absorbs the excitation light and transfers the energy to an “emitter” fluorophore which emits fluorescence. The same “harvester” fluorophore is used with various emitter fluorophores to generate multiple colors. Another possibility is to use quantum dots (QDs) with different emission wavelengths as the reporter dye. Similar to wavelength-shifting dyes, many QDs can be excited with a single UV lamp light source [22]. However, it remains to be seen if QDs can be effectively quenched and if their functional size can be reduced to be useful when conjugated to a MB.
Clearly, multicolor fluorescence detection of different beacon/target duplexes can become a powerful tool for the simultaneous detection of multiple genes. For example, almost all cancers are caused by multiple genetic alterations in cells and the detection of cancer cells in a clinical sample would require the use of multiple tumor markers. Thus, the use of multiple molecular beacons is potentially a powerful tool in the early detection and diagnosis of cancer.
Similar to fluorophore selection, choosing the optimal quencher can also improve the signal-to-background ratio of molecular beacons. Organic quencher molecules such as dabcyl, BHQ-IITM (blackhole quencher) (Biosearch Tech), BHQ-III (Biosearch Tech) and Iowa Black (IDT) can all effectively quench a wide range of fluorophores by both fluorescence resonance energy transfer (FRET) and the formation of an exciton complex between the fluorophore and the quencher [23]. In addition to organic quenchers, gold nanoparticles can also be used as quenchers [24]. It should be noted, however, that the interaction between the gold particle and the fluorophore could significantly affect the performance of molecular beacons.
12.3. IN VITRO GENE DETECTION
12.3.1. Pathogen Detection
The detection and identification of pathogens is often painstaking and fruitless due to the low abundance of diseased cells in clinical samples. The genomic sequences of the pathogen can be amplified through methods such as PCR and nucleic acid sequence-based amplification (NASBA), but the nucleic acid targets are often lost amidst other unintended products of amplification. The unique properties of molecular beacons have led to their application in a large number of homogeneous assays involving the sensitive detection of diseased states. Since most pathogens can be identified by their genomic sequences, molecular beacon-PCR assays provide a rapid and accurate method for pathogen detection and identification by simply adding molecular beacons to a typical PCR reaction tube. During
HAIRPIN NANOPROBES FOR GENE DETECTION |
409 |
the annealing stage, the molecular beacons bind to the desired target and a fluorescence signal is generated. Excess molecular beacons remain in the stem-loop structure and thus do not emit fluorescence. Therefore, the intensity of the signal is proportional to the target copy number. Further, when the temperature is increased during the extension stage, the molecular beacon melts away from the target and thus does not interfere with polymerization. It is important that the target-binding domain of the molecular beacon be designed with a melting temperature slightly above the annealing temperature for optimal results. The melting temperature of the stem should be about 10◦C above the annealing temperature to ensure that unbound molecular beacons remain in the stem-loop conformation and, for the most part, are not open due to thermal fluctuations. Since molecular beacons can detect complementary targets during PCR amplification, subsequent handling is not necessary, allowing the use of sealed tubes and reducing the risk of carry-over contamination. Further, since molecular beacons only fluoresce in the presence of complementary targets, unintended amplification products such as “primer-dimers” and false amplicons are not detected.
It is not hard to imagine the development of simple and rapid molecular beaconbased assays for the sensitive detection of nucleic acids in a clinic to help identify specific pathogens. Molecular beacons can easily be used to differentiate between fungal pathogens such as Candida dubliniensis and Candida albicans, which possess similar phenotypic and genotypic characteristics [25, 26]. In a controlled study, the correct pathogen was identified 100% of the time following PCR amplification. Such accurate determination of relevant disease states could provide improved strategies for proper disease management. In another example, NASBA utilized with molecular beacons was able to correctly identify West Nile virus, St. Louis encephalitis, and Hepatitis B viruses with high sensitivity and specificity [27, 28]. This type of assay can easily be exploited for clinical use as a common blood screening diagnostics test.
Molecular beacons have also been found to be useful as a fast and reliable tool for the timely detection of food and water-borne pathogens, such as Salmonella [29]. Molecular beacons successfully differentiated between Salmonella and similar pathogens such as Escherichia coli and Citrobacter freundii. Therefore, detection of such pathogens can prove to be helpful in preventing bacterial disease outbreaks. Molecular beacons or other nanostructured molecular probes also have the potential to become a powerful tool in biodefense.
Using molecular beacons for the accurate detection of nucleic acid targets is not, of course, limited to the identification of pathogens but has also been extended to other assays such as the determination of the sex of embryos [30], and the differential expression of specific genes under varying environmental conditions [31]. The potential applications of molecular beacons in pathogen detection seem limitless.
12.3.2. Mutation Detection and Allele Discrimination
One of the major benefits of molecular beacons is their ability to discriminate between targets with just a single base mismatch. The stem-loop structure of molecular beacons increases the specificity of beacon/target hybridization compared with linear probes and thus may offer advantages over other reporter probes such as Taqman (Applied Biosystems) [32]. Assays requiring the detection of single nucleotide polymorphisms can be performed with either one or two molecular beacons [33]. For single molecular beacon assays, the molecular beacon must be carefully designed such that during the annealing stage, it only
410 |
PHILIP SANTANGELO ET AL. |
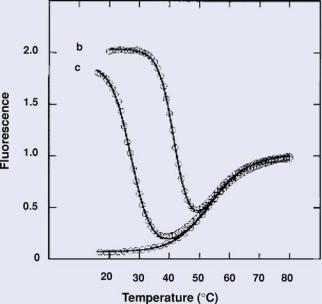
a
FIGURE 12.3. Thermal denaturation profiles of solutions containing molecular beacons: curve a, in the absence of targets; curve b, in the presence of a 6-fold excess of perfectly complementary targets; and curve c, in the presence of a 6-fold excess of single-base mismatched targets (Adapted from Ref. 16).
binds to the complementary target. This often requires the generation of thermal denaturation profiles to determine the “window of discrimination”, as illustrated in Figure 12.3 where the best assay temperature is at 37◦C. Specifically, an annealing temperature is determined that allows molecular beacons to hybridize to perfectly complementary targets but not targets with single-base mismatches. For assays utilizing two molecular beacons, one molecular beacon is designed to hybridize to the wild-type target, while the second molecular beacon is designed to hybridize to the mutant target. Each molecular beacon is labeled with a unique fluorophore with non-overlapping emission curves. Typically only the reporter fluorescence corresponding to the amplified target (wild-type or mutant) is detected. If both types of target are present and amplified, fluorescence from both probes will be detected. The competitive nature of this assay allows for high specificity and sensitivity. It has been found that even just 10 copies of a rare target can be detected in the presence of 100,000 copies of abundant target after PCR amplification [34]. A variant level of about 1% was detected following NASBA [35]. These levels of sensitivity are far superior to sequencing, which can only detect variant levels of about 10–20%.
The exact fraction of mutant alleles in a clinical sample/tissue can be determined by performing what has been dubbed “digital PCR” [36]. In this assay, the extracted DNA templates are diluted into multi-well plates such that there is only one template molecule per two wells, on average. After PCR, the fluorescent signal indicates whether the template was wild-type or mutant. The fluorescent signals from a microplate can therefore provide a digital readout of the fraction of mutant alleles. Recently, this assay has been successfully used
HAIRPIN NANOPROBES FOR GENE DETECTION |
411 |
to demonstrate the presence of allelic imbalance in colorectal tumors [37]. An alternative method to quantify heteroplasmy levels involves comparing the fluorescence intensities of the differently labeled molecular beacons during the cycle of maximum amplification [12]. This information can be obtained by taking the first derivative of the amplification curve and integrating over several of the cycles surrounding the cycle of maximum amplification. It was found that as little as 5% heteroplasmy could be measured reliably using this method.
The high sensitivity and single base specificity of molecular beacon-based assays has extended their use to many clinical and epidemiological applications. In one example, a clinical assay that detects point mutations was designed to determine whether or not Plasmodium falciparum samples, a parasite that causes malaria, contained an antifolate resistance-associated mutation [38]. Identification of such a mutation is extremely important for proper drug treatment. Assays have also been developed to rapidly screen blood samples for mutations in specific genes such as methylenetetrahydrofolate reductase (MTHFR) [39]. A cytosine to thymine mutation in MTHFR has been related to increased risk of cardiovascular disease and neural tube defects. The identification of such a mutation could lead to improved patient awareness. More information concerning the design and applications of molecular beacons can be found in www.molecular-beacons.org.
12.4. INTRACELLULAR RNA TARGETS
One of the most exciting and promising applications of nanostructured molecular probes such as molecular beacons is their potential use for the real-time visualization of RNA expression in living cells and tissues. For example, the ability to monitor the level of mRNA expression in living cells will provide important information concerning the temporal and spatial processing, localization, and transport of specific mRNA under various conditions. Further, detecting pathogenic markers will provide a means of locating and identifying diseased cells, allowing rapid diagnosis and prognosis of a disease.
To sensitively detect and quantify mRNA levels in live cells, it is extremely important to understand the form, distribution, and dynamics of target mRNA in live cells in order to optimize the probe design and measurement. Further, it is important to understand the impact of the probe on the cell, including its delivery, chemistry, possible toxicity, and nonspecific interactions. In the following sections, we describe the basic features of mRNA in living cells, aiming to set a stage for more discussions in Section 5 on intracellular mRNA detection and quantification.
12.4.1. Cytoplasmic and Nuclear RNA
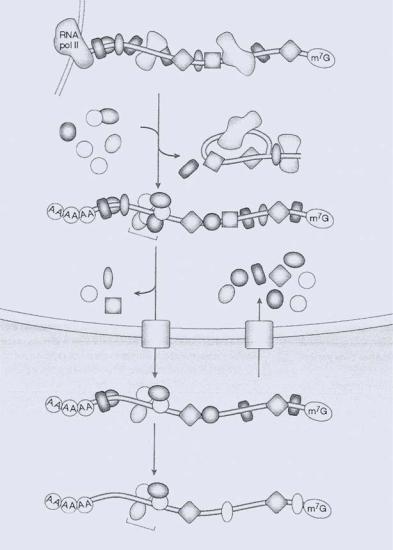
In a living cell, the functional forms of pre-mRNA/mRNA exist as ribonucleoprotein complexes (RNPs) in which numerous heterogeneous nuclear ribonucleoproteins (hnRNPs) bind to the transcript [40]. The association of these proteins begins during transcription, and there is evidence to show that some of these proteins remain bound to the mRNA all the way to the ribosome. The distribution of these proteins along the transcript, as well as the dynamic nature of the protein/RNA association, has a significant impact on the accessibility of pre-mRNA/mRNA to hairpin probes (see Figure 12.4).
412 |
PHILIP SANTANGELO ET AL. |
Chromatin
|
hnRNA (pre-mRNA)–hnRNP/snRNP |
|
magoh |
Y14 |
Splicing |
|
||
Aly/REF |
snRNP |
|
Upf3 |
||
|
||
RNPS1 |
|
|
SRm160 |
Excised intron |
|
|
||
EJC |
mRNA–hnRNP |
|
|
||
Nucleus |
|
|
|
mRNA export |
|
Cytoplasm |
|
|
|
hnRNP |
|
|
shuttle |
|
hnRNP/mRNP |
mRNA–hnRNP |
|
|
||
exchange |
|
PABP
cEJC |
mRNA–hnRNP |
|
FIGURE 12.4. Illustration of HnRNP proteins and mRNP proteins along the pathway of mRNA biogenesis, together with an overview of the interaction and dynamics of the myriad of RNA-binding proteins with premRNA and mRNA. EJC, exon-exon junction complex; NMD, nonsense-mediated mRNA decay; RNA pol II, RNA polymerase II; hnRNP, heterogeneous nuclear ribonucleoprotein; snRNP, small nuclear ribonucleoprotein; mRNP, mRNA-protein complex; PABP, poly(A)-binding protein; m7G, methylguanosine cap (Adapted from Ref 40).
12.4.1.1. Transcription and Polyadenylation mRNA is transcribed by a protein transcription complex containing RNA polymerase II which, especially the carboxy-terminal domain, couples transcription with mRNA processing [41]. All pre-mRNA processing is a co-transcriptional event, including addition of the poly(A) tail and splicing. Even as the nascent transcript (20–25 nucleotides in length) is emerging from the transcription complex,
HAIRPIN NANOPROBES FOR GENE DETECTION |
413 |
a capping enzyme binds the 5’ end, attaching the 5’-7-methylguanosine cap (reviewed in [42]. This cap is then methylated in order to stabilize the transcript against 5’ exonucleolytic attack [43]. The methylated end is also a signal for nuclear cap binding proteins that facilitate the interaction between the 5’ splice site of a pre-mRNA containing introns and a protein complex called the spliceosome. Depending upon a functional poly(A) signal and the terminal splice acceptor site of the terminal intron [44], the termination of transcription occurs far downstream of the poly(A) site, either before or at the same time as poly(A) is cleaved due to cleavage factors [45]. The addition of the 3’ poly(A) tail of a pre-mRNA requires more than a dozen polypeptides to be present [42]. The pre-mRNA first undergoes endonucleolytic cleavage by an endonuclease at the poly(A) synthesis initiation site, followed by processive poly(A) synthesis by a poly(A) polymerase. The poly(A) tail (ranging in length from 20 to 250 nucleotides) is then bound by poly(A) binding proteins [40].
12.4.1.2.Splicing During pre-mRNA processing, introns are removed from premRNA by a ribonucleoprotein (RNP) machine (spliceosome) that contains at least 50 proteins and 5 small nuclear RNAs [46]. This spliceosome is assembled at each intron, excising the intron and then releasing it in a branched “lariat” form along with the spliced pre-mRNA. Many of the splicing proteins remain bound to mRNA after splicing [47, 48], generating a specific nucleoprotein complex that facilitates mRNA export [49]. Specifically, some of the bound proteins are members of the SR (serine-arginine) protein family of splicing factors [49], a number of which have been shown to shuttle between the nucleus and cytoplasm [47]. Proteins within this complex (such as Y14 and Mago) can target an mRNA for nonsense-mediated mRNA decay or recruit proteins such as TAP/p15 to assist in nuclear export.
12.4.1.3.Nuclear Export and Localization Trans-acting factors (proteins that bind to cis elements or “zipcode” sequences within RNA) for mRNA localization are often present in granules (ribonuclear protein–RNP–complexes) that contain all components necessary for RNA processing, transport, localization, anchoring, and translation (reviewed in [50]. In other words, mRNA localization is a process initiated in the nucleus, based on the proteins that are bound during processing and accompany the mature mRNA from the nucleus to the cytoplasm. The hnRNP (heterogeneous nuclear ribonuclear protein) A/B family is one group of proteins that play a significant role in mRNA localization. Both hnRNP A1 and A2 leave the nucleus with mRNAs and then dissociate from the mRNA in the cytoplasm. Specifically, hnRNP A2 has been shown to help localize myelin basic protein (MBP) mRNA in oligodendrocytes [51–54]. In addition, it has roles in splicing, nuclear export, translational regulation, and RNA stabilization [50]. hnRNP A2 binds to a 21-nucleotide element in the 3’ untranslated region (UTR) of mRNA [53, 54]. A second example of a nuclear-bound trans-acting factor is zipcode-binding protein 2 (ZBP2), which binds to the 3’ UTR of β-actin mRNA [55].
The Tap protein is another protein that plays a significant role in nuclear RNA export [56–58]. This protein contains an RNA binding domain, a nuclear export signal, and a binding domain for nucleoporins (proteins that make up the nuclear pore) (reviewed in
[59].The recruitment of this protein to spliced mRNA has been proposed to be the final step required before nuclear pore binding [59].

414 |
PHILIP SANTANGELO ET AL. |
TABLE 12.1. Summary of proteins bound to pre-mRNA.
Type of Protein |
Description and function |
|
|
RNA polymerase II |
|
Capping enzyme |
|
Cap binding proteins |
May stay attached all the way through export from nucleus [42] |
Polyadenylation machinery |
At least 12 proteins including an as-of-yet unidentified endonuclease and |
|
Poly(A) polymerase, along with many other required protein factors. |
Poly(A) binding proteins |
|
Spliceosome |
A ribonucleoprotein (RNP) machine that contains at least 50 proteins and 5 |
|
small nuclear RNAs. Some of these proteins remain bound after splicing and |
|
are required for nuclear export [49]. |
Exon-exon junction complex |
A multi-protein complex 20–24 nucleotides upstream of an exon-exon junction |
|
[40, 60]. This complex binds to as little as eight nucleotides. |
hnRNP proteins |
An assortment of at least 20 proteins that associate with nascent pre-mRNA; |
|
has roles in localization, processing and nuclear transport. The binding sites |
|
of some of these proteins have been identified as being in the 3’ UTR of |
|
mRNAs. Others (of the hnRNP C family) associate preferentially with |
|
introns. |
|
|
12.4.1.4. Distribution of RNA Between Nucleus and Cytoplasm An important issue pertaining to the measurement of mRNA levels using hairpin probes is the relative levels of RNA in the nucleus and cytoplasm. It has been reported that a large fraction of RNA (>95%) synthesized by RNA polymerase II never leaves the nucleus as mature mRNA [61]. Further, over 1/3 of the RNAs never reaches the cytoplasm due to mRNA processing events (removal of introns, transcription termination). There is also a population of primary transcripts that are not polyadenylated or transported from the nucleus (termed “nonproductive hnRNAs”), and therefore never destined to produce mRNA [61]. It is therefore possible for probes to bind to mRNAs in the nucleus that may never be translated.
12.4.1.5. Transport and Localization of mRNP Following export from the nucleus, a mRNP is often transported to specific regions within cells. It is believed that the specific localization of mRNPs has a key role in the compartmentalization of protein synthesis in the cytoplasm [62–64]. Key questions remain open in this area include: (1) Are mRNPs localized in cells? If so, is there a general cell structure where most of the mRNPs are localized? (2) What is the intracellular system along which they are being transported to their destination? (3) What factors decide specific localization of mRNPs? (4) What is the biological significance of mRNP localization in cells?
Most mRNPs are believed to be associated with the cytoskeleton, which may be used to transport and localize the mRNP to specific sites within a cell. Although the details of this process remain elusive, there is growing evidence suggesting the importance of the cytoskeleton in mRNA localization. The key evidence is that there exists a close association of polyribosomes (also referred to as polysomes) with the cytoskeleton [65–68]. In addition, studies have shown that microtubules are involved in the assembly of membrane-bound polyribosomes [69, 70]. Using drug treatment to depolymerize microtubules, membranebound ribosomes were prevented from initiating protein synthesis and showed a decreased level of total poly(A) mRNP and fibronectin mRNP. Although these studies did not identify
HAIRPIN NANOPROBES FOR GENE DETECTION |
415 |
the nature of the interaction between membrane (ER) bound polysomes and microtubules, they did show a close relationship between the cytoskeleton, ER bound polysomes, mRNP transport and protein synthesis. Further, using both in vitro reconstitution and biochemical fractionation, Hamill et al [71] provided EM evidence that a fraction of polyribosomes/mRNPs was bound to microtubules.
12.4.1.6.Localization of mRNPs At present, there is no clear consensus as to what fraction of mRNPs are localized with the cytoskeleton. Biochemical evidence provided by [72] suggests that 70–80% of mRNPs in a cell are co-localized with the cytoskeleton. Due to the lack of fluorescent or chemical tags, the verification of the biochemical results in intact cells has been limited to one or two specific mRNPs. For example, using in situ hybridization and biochemical fractionation, Wiseman and Hesketh [73, 74] and Russell and Dix [75] have demonstrated co-localization of mRNPs of the myosin heavy chain with cytoskeletal elements. [65, 66, 76] have shown the association of actin and poly(A) mRNPs with cytoskeletal elements, especially microtubules. Using a similar in situ approach, [77] have implicated cytoskeletal localization of metallothionein I. There is a clear need to better understand the localization of mRNPs, their association with specific elements in a cell and the biological significance of co-localization.
It has been shown [77, 78] that the 3’ UTR plays a significant role in specific localization of certain mRNPs in cells. Specifically, using genetic engineering to perturb mRNA in the 3’ UTR region, it has been shown that the 3’ UTR regions of heavy chain myosin, actin, c-myc, metallothionein-I are critical for the localization of these specific mRNPs. Although the localization signal in the mRNA sequence has been identified, the structural basis of the 3’ UTR, the associated binding proteins and, perhaps more importantly, the processes involved in specific localization and transport are still poorly understood. To date, proteins responsible for the specific localization of mRNA have been identified only for oskar and nanos mRNA in Drosophila oocytes [79, 80] and for actin mRNA in chicken fibroblasts
[81].In both cases, cytoskeletal elements have been shown to be involved in transport and localization of the mRNPs. See Figure 12.5a and 12.5b for proposed roles of cytoskeleton in transport and localization of mRNP.
As mentioned earlier, it is mRNP rather than mRNA alone that provides the functional unit for localized protein synthesis. In order to understand the intracellular transport and localization of mRNA, extensive studies need to be performed to track the movement of mRNP complexes. Possible approaches for doing so include labeling proteins with GFP [82], generating transgenes with high affinity binding sites for a GFP tagged proteins [80], sitespecific protein labeling using FLAsH [83], and targeting mRNAs using molecular beacons
[84].These approaches in combination with high-resolution microscopy [85] or EM may reveal the detailed structural organization of mRNP transport and localization in living cells.
12.4.1.7.Degradation of mRNA One of major pathways of regulating posttranscriptional gene expression is the degradation of mRNA. Considerable focus has been placed on understanding the transcriptional controls of mRNA synthesis, but there is very limited understanding of the mechanisms responsible for controlling the rate of mRNA degradation, which affects the intracellular mRNA level. It has been suggested that a variety of physiological signals such as hormones [86, 87], iron binding proteins and cell cycle regulators
[88]may have significant effects on the decay rates of specific mRNAs, and the half-life of