

Micro-Nano Technology for Genomics and Proteomics BioMEMs - Ozkan
.pdf426 |
PHILIP SANTANGELO ET AL. |
class of nucleases is DNase II, which is an acid nuclease. The activity of all these nucleases is tightly regulated. CAD, for example, becomes active only after caspase-mediated cleavage of an inhibitory protein. DNase I is activated upon proteolytic cleavage from an inactive precursor. DNase II, a lysosomal acid hydrolase, is most active at low pH values.
Endo-exonucleases are Ca2+/Mg2+-dependent and proteolytically-activated; they may also play a role in apoptosis [137]. Endo-exonucleases primarily degrade single-stranded DNA, and exist in an inactive form in both the ER and cytosol. The active form was found primarily in the nuclei. Two precursor pools (ER and cytosolic) were proposed for the nuclease. The cytosolic pool is assumed to be involved in normal DNA repair and possibly in the nicking of supercoiled chromatin DNA. The ER-membrane bound nucleases are proposed to be a storage reserve for the enzyme to be mobilized in apoptosis. The common features of all DNAses are that their activation is usually tightly regulated by divalent ion concentration and pH and, once activated, they often translocate to the nucleus. Therefore, if the hairpin probes can be delivered through a non-endocytic pathway, DNase activation and thus probe degradation in the cytoplasm may be avoided.
12.5.2.2. RNases A wide variety of RNases have been identified that hydrolyze single-stranded RNA, double-stranded RNA, and RNA-DNA hybrid. RNases are typically concentrated in a few subcellular compartments and play different roles in RNA processing and metabolism. A specific family of RNases is a complex of 10 to 11 proteins termed “exosome” which has 3’ to 5’ exonuclease activity in both the nucleus and cytoplasm [138–140]. Quite a few, if not all, of the exosome proteins are exonucleases. There does not appear to be a substantial pool of free exosomal subunits; almost all the associated exonucleases in the cell are components of exosomes, even though they are all active exonucleases when isolated individually and assayed in vitro [140–142]. In the nucleus, the exosome has been shown to process 3’-extended ribosomal RNA. Exosomes may also play a role in nuclear mRNA degradation [143], and the degradation of pre-rRNA transcripts that are unable to be properly processed [144]. In the cytoplasm, the exosome is required for the 3’ to 5’ degradation of some poly(A) mRNAs, which may contribute to antiviral defense [145, 146]. Moore [139] proposed that this assembly of multiple exonucleases allows for both coordinated regulation and delivery of a variety of exonucleases. However, it is still unclear what controls the recognition of a specific substrate, although it has been suggested that some members of the RNA helicase family may play an important role in substrate recognition.
Acid ribonucleases (reviewed in [147, 148]) from the RNase T2 family degrade singlestranded RNA and are primarily localized in lysosomes, although members of the family have also been identified in the nucleus. The RNase A family of ribonucleases functions as antibacterial, anti-parasitic, and anti-viral agents [149], degrading single-stranded RNA. They are secretive proteins thus posing little, if any, threat to hairpin probes inside a cell.
RNase H is an enzyme that endonucleolytically degrades the RNA component of an RNA-DNA heteroduplex. Although the exact cellular function of RNase H enzymes is not fully understood, it has been suggested that Type 1 RNase H proteins are involved in processing RNA/DNA hybrids, whereas Type 2 RNase H enzymes are responsible for the removal of Okazaki primers during lagging strand DNA replication [150, 151]. It has also been demonstrated that Type 2 RNase H enzymes may play a role in the repair of misincorporated ribonucleotides [152]. The RNase H enzymes play a key role in the mechanism of inhibition of gene expression by antisense oligonucleotides. The catalytic activity of RNase
HAIRPIN NANOPROBES FOR GENE DETECTION |
427 |
H2 in particular is associated with antisense-mediated RNA degradation [153]. It was revealed that the expression levels of both RNase H1 and RNase H2 have large variations in different human cell lines [153]. Examination of the localization of green fluorescent protein (GFP)-tagged RNase H1 and RNase H2 indicated that in most cell types, RNase H1 was distributed throughout the whole cell whereas RNase H2 was only detectable in the nucleus.
As with DNases, RNases are tightly regulated, with specific substrates and conditions necessary for activation. However, RNases are not completely characterized, so their effect on hairpin probes is difficult to predict. It is believed that within the cytosol, outside the endocytotic pathway, few, if any, RNases will pose a threat to hairpin probes. However, the nuclear environment is much more complex, and nucleases in the cell nucleus are likely to impede the use of hairpin probes in mRNA detection.
12.5.2.3. Backbone Modifications The backbone chemistry of a hairpin probe has profound implications for the behavior of the probe in the intracellular environment. For example, chemically modified oligonucleotide backbones affect probe affinity, melting temperatures, and nuclease resistance. Extensive studies of backbone modification have been performed to enhance nuclease resistance. The most common modification of the antisense probes is to replace the phosphodiester bond with a phosphorothioate bond in the oligonucleotide. In phosphorothioate oligonucleotides (PS-ODNs), a sulfur atom replaces one of the non-bridging oxygen atoms in the phosphate backbone. This modification has been shown to have greater resistance to nuclease degradation than phosphodiesters while maintaining RNase H-mediated mRNA degradation [reviewed in 154]. The melting temperature of the PS- ODN-target RNA duplex is slightly lower than the corresponding phosphodiester oligonucleotide duplex and PS-ODNs have lower affinities than the unmodified DNA oligonucleotides for the RNA target [155]. A major drawback of this backbone modification is its polyanionic backbone, which can lead to elevated protein binding and immunoresponse [154]. Excess protein binding would certainly be detrimental to the ability of a hairpin probe to hybridize to its target mRNA. It may also activate RNase H activity, which is desirable for antisense therapy, but not desirable when the probes are used for imaging mRNA expression in living cells.
Modification of the sugar moiety of an oligonucleotide forms the basis for another class of modified oligonucleotides. A number of modifications have been made at the 2’ position of the sugar ring [reviewed in 156]. Adding a 2’-O-methyl group at this position significantly increases RNA binding affinity and enhances nuclease resistance. 2’-O-methyl backbone modification results in an increase in melting temperature on the order of 1◦C per modified oligonucleotide [157]. Although this modification is not ideal for antisense therapy because it is RNase H-resistant (the probe-target hybrid is more an RNA:RNA duplex than a DNA:RNA duplex that RNase H recognizes), it is beneficial for a hairpin probe in that it helps avoid RNase H-mediated RNA degradation. 2’-O-methyl modified molecular beacons, however, may be more prone to opening by hairpin binding proteins. It may also trigger unwanted RNA interference.
Another nucleic acid analog in which the sugar ring is modified has been described as “locked nucleic acid” [LNA, reviewed in 158]. In this backbone modification, the 2’-O and 4’-C are linked by a methylene linker. This “locks” the sugar into the conformation
428 |
PHILIP SANTANGELO ET AL. |
(C-3’-endo sugar) that is supposedly ideal for recognition of RNA. This modification results in significant increases in melting temperatures (ranging from 1◦C to 8◦C against DNA) and has higher specificity than unmodified DNA oligonucleotides. LNAs have higher affinity for targets primarily due to slower dissociation, as shown by stopped-flow kinetics experiments [159]. LNAs have also been shown to be stable in the presence of a number of nucleases [160].
Peptide nucleic acids (PNAs) are nucleotide analogs that also offer advantages for the design of hairpin probes. PNA is a DNA mimic where the phosphodiester-linked backbone is replaced with an N-(2-aminoethyl)glycine backbone [161] and behaves according to traditional Watson-Crick base-pairing rules. PNAs have very high binding affinity for singlestranded DNA and RNA; the melting temperature is increased by 1.45◦C per monomeric PNA unit [155]. The rate of hybridization is at least as fast as DNA:DNA duplex formation. PNA:RNA duplexes are also more stable at lower salt concentrations due to the neutral character of the PNA backbone. PNA oligonucleotides exhibit both high affinity and high specificity [162] when binding to DNA. PNA oligonucleotides are completely nucleaseresistant. They also have the unique ability to “invade” a DNA duplex, although this strand displacement occurs only at salt concentrations much lower than the physiologically relevant value [reviewed in 155]. A careful study of the ability of PNA probes to bind to structured targets (i.e., those with hairpin structural motifs) showed that the higher affinity conferred by the PNA backbone enhances probe binding over a natural DNA probe [163], suggesting that PNA hairpin probes may have a distinct advantage when targeting an mRNA sequence ‘buried’ in the secondary and tertiary structure.
Optimization of hairpin probe design and performance requires the examination of different backbone modifications, including their combinations. Antisense research has shown that partial backbone modifications (i.e., modifying only a few oligonucleotide bases) often confer desired properties. A combination of different modifications may lead to the ideal probe backbone for a given target. One target sequence, for example, may exhibit significant secondary structure, while another may be occluded by a number of bound proteins. Hybridization to a specific target sequence, therefore, may require a specific backbone modification.
12.5.3. Intracellular mRNA Detection
One of the major challenges in measuring endogenous gene expression in living cells and tissue is the design of reporter probes with high sensitivity, specificity and signal-to- background ratio. In addition to challenges in cellular delivery and maintaining integrity of probe in cell cytoplasm, it is critical to have good target accessibility, which is largely controlled by mRNA secondary/tertiary structures and RNA-binding proteins. Specifically, an mRNA molecule in a cell usually has a folded conformation with double stranded segments. Further, mRNAs almost always have proteins bound to it, which may alter mRNA structure and prevent probe binding. Therefore, in selecting the probe sequences, it is important to avoid targeting sequences that are ‘buried’ inside the tertiary structure or where double stranded RNA is formed. Although predictions of mRNA secondary structure can be made using existing software, they may not be very accurate due to limitations of the biophysical models used. Further, we only have very limited knowledge of the sequences occupied dynamically by RNA-binding proteins. Therefore, for each gene to target, it may be necessary to select multiple unique sequences along the target RNA, and
HAIRPIN NANOPROBES FOR GENE DETECTION |
429 |
|
|
|
|
|
|
|
|
|
|
|
Acceptor |
|
|
|
|
|
|
|
|
|
|
|
|
|
Molacular |
|
|
|
|
|
|
|
|
Donor |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
Molacular |
|
|
|
Beacon |
|
||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
Beacon |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quencher |
|
|
|
|
|
FRET |
Quencher |
|||||
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
Acceptor Dye |
||||
|
|
|
|
|
|
|
Donor Dye |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
mRNA Target |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
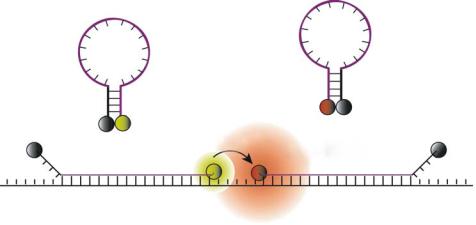
FIGURE 12.8. A schematic illustration showing the concept of dual FRET molecular beacons. Hybridization of donor and acceptor molecular beacons to adjacent regions on the same mRNA target results in FRET between donor and acceptor fluorophores upon donor excitation. By detecting FRET signal, fluorescence signals due to probe/target binding can be readily distinguished from that due to molecular beacon degradation and non-specific interactions (Adopted from Ref 84).
have corresponding molecular beacons designed, synthesized and tested in cells to achieve high signal-to-background ratio. Clearly, a better understanding of mRNA structure and RNA-protein interactions will facilitate significantly the design of hairpin probes for gene detection.
To drastically reduce the false-positive signals, two different approaches have been taken. The first is a dual molecular beacons approach which measures the fluorescent signal due to fluorescence resonance energy transfer (FRET) or luminescence resonance energy transfer (LRET) as a result of the direct interaction between two molecular beacons [18, 84, 117]. The second approach is to modify the backbone of molecular beacons to make them more resistant to endogenous nucleases, as discussed earlier. For certain applications such as long time monitoring of mRNA expression in living cells, it may require the combination of both approaches in order to achieve high signal-to-background ratio.
As shown in Figure 12.8, the dual FRET/LRET molecular beacons approach utilizes a pair of molecular beacons, one with a donor fluorophore and a second with an acceptor fluorophore. Probe sequences are chosen such that the molecular beacons hybridize adjacent to each other on a single nucleic acid target, bringing the respective fluorophores into close proximity and promoting FRET [18, 84]. The sensitized emission from the acceptor fluorophore then serves as a positive signal in the FRET based detection assay. When the donor and acceptor fluorophores are properly chosen (e.g., with minimal spectral overlap), a strong fluorescence signal will emit from the acceptor only when both molecular beacons are hybridized to the same target and FRET occurs. The fluorescence emitted from molecular beacons that are degraded or opened by protein interactions will be substantially lower than the signal elicited by the donor/acceptor FRET interaction. Thus, a true positive signal owing to probe/target binding events can be readily distinguished from false-positive signals.
To demonstrate the potential of dual FRET molecular beacons approach, an in-solution spectroscopy study was carried out [18]. Specifically, a series of molecular beacons were
HAIRPIN NANOPROBES FOR GENE DETECTION |
431 |
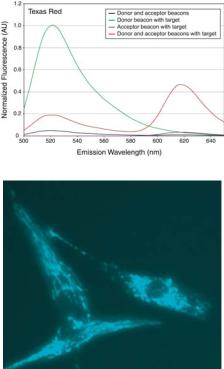
was used as the donor dye and Texas Red (perk emission at 620 nm) as the acceptor dye, a signal-to-background ratio of about 50 was achieved. Clearly, the FRET signal should allow differentiation between the signal emitted upon target detection and false-positive signals.
When the distance between the donor and acceptor molecular beacons was increased from 3 to 6 bases, there was a slight increase in the FRET signal intensity (data not shown). This trend was found to be the same for all the acceptor fluorophores studied. It seems that a gap size of 4 or 5 bases is desirable whereas a 3-base gap is unfavorable due to possible interference between the donor and acceptor dyes.
To demonstrate the ability of molecular beacons in sensitive detection of mRNA in living cells, dual-FRET molecular beacons targeting wild-type K-ras mRNA were designed and synthesized; they were delivered into normally-growing and stimulated human dermal fibroblasts (HDF cells) using SLO. A control molecular beacon (random beacon) was also designed whose sequence has no perfect match in the mammalian genome. Both the K-ras-targeting dual FRET molecular beacon pair, and control molecular beacons have a 16-base target sequence and 5-base stem [84]. For both molecular beacon pairs, Cy3 and Cy5 fluorophores were used as the donor and acceptor, respectively. One hour after the delivery of molecular beacons using SLO, the HDF cells were excited at 545 nm (Cy3 maximum excitation), and the resulting fluorescence signal was observed at 665 nm (Cy5 maximum emission). It was found that the dual-FRET molecular beacons approach could provide fascinating images of mRNA localization [84]. As an example, Fig. 12.9b shows a fluorescence image of K-ras mRNA localization in a few stimulated HDF cells. This direct visualization of mRNA in single living cells revealed intriguing filamentous localization pattern. Evidently, K-ras mRNA molecules are not randomly distributed in the cytoplasm, but localized, possibly to a cytoskeletal component. It is very important to understand why K-ras mRNAs localize in such a way, and what are the biological implications. It is also essential to study the localization of mRNAs corresponding to different classes of proteins. It is likely that the molecular beacons approach will provide essential information on mRNA synthesis, processing, transport, localization, and dynamics in living cells.
12.6. OPPORTUNITIES AND CHALLENGES
Nanostructured molecular probes such as molecular beacons have the potential to enjoy a wide range of applications that require rapid and sensitive detection of genomic sequences. However, to date molecular beacons are used mostly as a tool for the detection of single stranded nucleic acids in homogeneous in vitro assays. For example, molecular beacons have been modified for solid phase studies [164, 165]. Surface immobilized molecular beacons used in microarray assays allow for the high throughput parallel detection of nucleic acid targets while avoiding the difficulties associated with PCR-based labeling [164, 166]. Another novel application of molecular beacons is the detection of double-stranded DNA targets using PNA “openers” that form triplexes with the DNA strands [19]. Further, proteins can be detected by synthesizing “aptamer molecular beacon” [167, 168] which, upon binding to a protein, undergoes a conformational change that results in the restoration of fluorescence.
Perhaps the most exciting application of hairpin probes including molecular beacons is living cell gene detection. As demonstrated, the dual FRET molecular beacons technique
432 |
PHILIP SANTANGELO ET AL. |
can detect endogenous mRNA in living cells rapidly with high specificity, sensitivity, and signal-to-background ratio, thus providing a powerful tool for laboratory and clinical studies of gene expression in vivo. For example, in drug discovery, this method can be used in highthroughput assays to quantify and monitor the dose-dependent changes of specific mRNA expression in response to different candidate drug molecules. In basic biological studies, this method will allow real-time visualization of the dynamics and localization of specific RNAs. The ability to detect and quantify the expression of specific genes in living cells in real-time will offer tremendous opportunities for biological and disease studies, provide another leap forward in our understanding of cell and developmental biology, disease pathophysiology, and significantly impact medical diagnostics.
There are a number of challenges in detecting and quantifying RNA expression in living cells. In addition to probe design issues and target accessibility, quantification of mRNA expression in single cells poses a significant challenge. For example, it is necessary to distinguish true and background signals, determine the fraction of mRNA molecules hybridized with probes, and quantify the possible self-quenching effect of the reporter, especially when mRNA is highly localized. Since the fluorescence intensity of the reporter may be altered by the intracellular environment, it is also necessary to create an internal control by, for example, injecting fluorescently labeled oligonucleotides with known quantity into the same cells and obtaining the corresponding fluorescence intensity. Further, unlike in RT-PCR studies where the mRNA expression is averaged over a large number of cells (usually over one million), in optical imaging of mRNA expression in living cells, only a relatively small number of cells (typically less than one thousand) are observed. Therefore, the average copy number per cell may change with the total number of cells observed due to the (often large) cell-to-cell variation of mRNA expression.
Binding between proteins and hairpin probes may cause non-specific opening of the hairpin, thus increase the background signal. There are many proteins that could bind nucleic acids in the cell cytoplasm, especially hairpin-binding proteins. Although certain modifications of the oligonucleotide backbone chemistry may help, other modifications, such as 2’-O-metheyl chemistry, may increase the likelihood of probes being recognized by hairpin-binding proteins.
Another issue in living cell gene detection using hairpin ODN probes is the possible effect of probes on normal cell function, including protein expression. As revealed in the antisense therapy research, complementary pairing of a short segment of an exogenous oligonucleotide to mRNA can have a profound impact on protein expression levels and even cell fate. For example, tight binding of the probe to the translation start site can block mRNA translation. Binding of a DNA probe to mRNA can also trigger RNase H-mediated mRNA degradation. However, the probability of eliciting antisense effects with hairpin probes may be very low when low concentrations of probes (<200 nM) are used for mRNA detection, in contrast to the high concentrations (typically 20 µM; [120]) employed in antisense experiments. Further, it generally takes 4 hours before any noticeable antisense effect occurs, whereas visualization of mRNA with hairpin probes requires less than 2 hours after delivery. However, it is important to carry out a systematic study of the possible antisense effects. When 2’-O-metheyl hairpin probes are used, hybridization between the RNA-like probe and mRNA target results in double-stranded RNA, which may trigger RNA interference. This possibility should also be investigated.
HAIRPIN NANOPROBES FOR GENE DETECTION |
433 |
ACKNOWLEGEMENT
This work was supported in part by NSF (BES-0222211), and by DARPA/AFOSR (F49620-03-1-0320).
REFERENCES
[1]F.H. Crick. Symp. Soc. Exp. Biol., 12:138–163, 1958.
[2]B.D. Hall and S. Spiegelman. Proc. Natl. Acad. Sci. U.S.A., 47:137–163, 1961.
[3]J.D. Watson and F.H. Crick. Nature, 171:737–738, 1953.
[4]J. Hachmann and H.G. Khorana. J. Am. Chem. Soc., 91:2749–2757, 1969.
[5]R.K. Saiki, S. Scharf, F. Faloona, K.B. Mullis, and G.T. Horn et al. Science, 230:1350–1354, 1985.
[6]J.C. Alwine, D.J. Kemp, B.A. Parker, J. Reiser, and J. Renart et al. Methods Enzymol., 68:220–242, 1979.
[7]M.D. Adams, M. Dubnick, A.R. Kerlavage, R. Moreno, J.M. Kelley et al. Nature, 355:632–634, 1992.
[8]V.E. Velculescu, L. Zhang, B. Vogelstein, and K.W. Kinzler. Science, 270:484–487, 1995.
[9]P. Liang and A.B. Pardee. Science, 257:967–971, 1992.
[10]M. Schena, D. Shalon, R.W. Davis, and P.O. Brown. Science, 270:467–470, 1995.
[11]H.A. Erlich, D. Gelfand, and J.J. Sninsky. Science, 252:1643–1651, 1991.
[12]K. Szuhai, E. Sandhaus, S.M. Kolkman-Uljee, M. Lemaitre, and J.C. Truffert et al. Am. J. Pathol., 159:1651– 1660, 2001.
[13]M. Buongiorno-Nardelli and F. Amaldi. Nature, 225:946–948, 1970.
[14]S. Tyagi and F.R. Kramer. Nat. Biotechnol., 14:303–308, 1996.
[15]A. Tsourkas, M.A. Behlke, S.D. Rose, and G. Bao. Nucleic Acids Res., 31:1319–1330, 2003.
[16]G. Bonnet, S. Tyagi, A. Libchaber, and F.R. Kramer. Proc. Natl. Acad. Sci. U.S.A., 96:6171–6176, 1999.
[17]D.J. States, W. Gish, and S.F. Altschul. Methods, 3:66–70, 1991.
[18]A. Tsourkas, M.A. Behlke, Y. Xu, and G. Bao. Anal. Chem., 75:3697–3703, 2003.
[19]H. Kuhn, V.V. Demidov, J.M. Coull, M.J. Fiandaca, B.D. Gildea, and M.D. Frank-Kamenetskii. J. Am. Chem. Soc., 124:1097–1103, 2002.
[20]S. Tyagi, D.P. Bratu, and F.R. Kramer. Nat. Biotechnol., 16:49–53, 1998.
[21]S. Tyagi, S.A. Marras, and F.R. Kramer. Nat. Biotechnol., 18:1191–1196, 2000.
[22]W.C. Chan, and S. Nie. 1998 Science, 281:2016–2018.
[23]S.A. Marras, F.R. Kramer, and S. Tyagi. Nucleic Acids Res., 30:e122, 2002.
[24]B. Dubertret, M. Calame, and A.J. Libchaber. Nat. Biotechnol., 19:365–370, 2001.
[25]A. Sebti, T.E. Kiehn, D. Perlin, V. Chaturvedi, and M. Wong et al. Clin. Infect. Dis., 32:1034–1038, 2001.
[26]S. Park, M. Wong, S.A. Marras, E.W. Cross, T.E. Kiehn et al. J. Clin. Microbiol., 38:2829–2836, 2000.
[27]S. Yates, M. Penning, J. Goudsmit, I. Frantzen, B. van de Weijer et al. J. Clin. Microbiol., 39:3656–3665, 2001.
[28]R.S. Lanciotti and A.J. Kerst. J. Clin. Microbiol., 39:4506–4513, 2001.
[29]W. Chen, G. Martinez, and A. Mulchandani. Anal. Biochem., 280:166–172, 2000.
[30]K.E. Pierce, J.E. Rice, J.A. Sanchez, C. Brenner, and L.J. Wangh. Mol. Hum. Reprod., 6:1155–1164, 2000.
[31]R. Manganelli, E. Dubnau, S. Tyagi, F.R. Kramer, and I. Smith. Mol. Microbiol., 31:715–724, 1999.
[32]I. Tapp, L. Malmberg, E. Rennel, M. Wik, and A.C. Syvanen. Biotechniques, 28:732–738, 2000.
[33]M.M. Mhlanga and L. Malmberg. Methods, 25:463–471, 2001.
[34]J.A. Vet, A.R. Majithia, S.A. Marras, S. Tyagi, and S. Dube et al. 1999. Proc. Natl. Acad. Sci. U.S.A., 96:6394–6399.
[35]A. de Ronde, M. van Dooren, L. van Der Hoek, D. Bouwhuis, and E. de Rooij et al. J. Virol., 75:595–602, 2001.
[36]B. Vogelstein and K.W. Kinzler. Proc. Natl. Acad. Sci. U.S.A., 96:9236–9241, 1999.
[37]I.M. Shih, W. Zhou, S.N. Goodman, C. Lengauer, K.W. Kinzler, and B. Vogelstein. Cancer Res., 61:818– 822, 2001.
434 |
PHILIP SANTANGELO ET AL. |
[38]R. Durand, J. Eslahpazire, S. Jafari, J.F. Delabre, and A. Marmorat-Khuong et al. Antimicrob. Agents Chemother., 44:3461–3464, 2000.
[39]B.A. Giesendorf, J.A. Vet, S. Tyagi, E.J. Mensink, F.J. Trijbels, and H.J. Blom. Clin. Chem., 44:482–486, 1998.
[40]G. Dreyfuss, V.N. Kim, and N. Kataoka. Nat. Rev. Mol. Cell. Biol., 3:195–205, 2002.
[41]S. McCracken, N. Fong, K. Yankulov, S. Ballantyne, and G. Pan et al. Nature, 385:357–361, 1997.
[42]A.J. Shatkin and J.L. Manley. Nat. Struct. Biol., 7:838–842, 2000.
[43]Y. Furuichi and A.J. Shatkin. Adv. Virus. Res., 55:135–184, 2000.
[44]M.J. Dye and N.J. Proudfoot. Mol. Cell., 3:371–378, 1999.
[45]C.E. Birse, L. Minvielle-Sebastia, B.A. Lee, W. Keller, and N.J. Proudfoot. Science, 280:298–301, 1998.
[46]C.A. Collins and C. Guthrie. Nat. Struct. Biol., 7:850–854, 2000.
[47]J.F. Caceres, G.R. Screaton, and A.R. Krainer. Genes. Dev., 12:55–66, 1998.
[48]H. Le Hir, M.J. Moore, and L.E. Maquat. Genes. Dev., 14:1098–1108, 2000.
[49]M.J. Luo, and R. Reed. Proc. Natl. Acad. Sci. U.S.A., 96:14937–14942, 1999.
[50]K.L. Farina and R.H. Singer. Trends Cell. Biol., 12:466–472, 2002.
[51]K. Kristensson, N.K. Zeller, M.E. Dubois-Dalcq, and R.A. Lazzarini. J. Histochem. Cytochem., 34:467–473, 1986.
[52]A.N. Verity and A.T. Campagnoni. J. Neurosci. Res., 21:238–248, 1988.
[53]K. Ainger, D. Avossa, A.S. Diana, C. Barry, E. Barbarese, and J.H. Carson. J. Cell. Biol., 138:1077–1087, 1997.
[54]K.S. Hoek, G.J. Kidd, J.H. Carson, and R. Smith. Biochemistry, 37:7021–7029, 1998.
[55]W. Gu, F. Pan, H. Zhang, G.J. Bassell, and R.H. Singer. J. Cell. Biol., 156:41–51, 2002.
[56]J. Katahira, K. Strasser, A. Podtelejnikov, M. Mann, J.U. Jung, and E. Hurt. Embo. J., 18:2593–2609, 1999.
[57]A.E. Pasquinelli, R.K. Ernst, E. Lund, C. Grimm, M.L. Zapp et al. Embo. J., 16:7500–7510, 1997.
[58]C. Saavedra, B. Felber, and E. Izaurralde. Curr. Biol., 7:619–628, 1997.
[59]B.R. Cullen. Proc. Natl. Acad. Sci. U.S.A., 97:4–6, 2000.
[60]I.M. Palacios. Curr. Biol., 12:R50–R52, 2002.
[61]D.A. Jackson, A. Pombo, and F. Iborra. Faseb J., 14:242–254, 2000.
[62]G. Aakalu, W.B. Smith, N. Nguyen, C. Jiang, and E.M. Schuman. Neuron, 30:489–502, 2001.
[63]E.A. Shestakova, R.H. Singer, and J. Condeelis. Proc. Natl. Acad. Sci. U.S.A., 98:7045–7050, 2001.
[64]J.E. Hesketh. Exp. Cell. Res., 225:219–236, 1996.
[65]G.J. Bassell, R.H. Singer, and K.S. Kosik. Neuron, 12:571–582, 1994.
[66]G. Bassell and R.H. Singer. Curr. Opin. Cell. Biol., 9:109–915, 1997.
[67]J. Hesketh, D. Jodar, A. Johannessen, K. Partridge, I. Pryme, and A. Tauler. Biochem. Soc. Trans., 24:187S, 1996.
[68]P. Mahon, K. Partridge, J.H. Beattie, L.A. Glover, and J.E. Hesketh. Biochim. Biophys. Acta., 1358:153–162, 1997.
[69]P.R. Walker and J.F. Whitfield. J. Biol. Chem., 260:765–770, 1985.
[70]B. Zhou and M. Rabinovitch. Circ. Res., 83:481–489, 1998.
[71]D. Hamill, J. Davis, J. Drawbridge, and K.A. Suprenant. J. Cell. Biol., 127:973–984, 1994.
[72]S.K. Pramanik, R.W. Walsh, and J. Bag. Eur. J. Biochem., 160:221–230, 1986.
[73]J.W. Wiseman, L.A. Glover, and J.E. Hesketh. Biochem. Soc. Trans., 24:188S, 1996.
[74]J.W. Wiseman, L.A. Glover, and J.E. Hesketh. Cell. Biol. Int., 21:243–248, 1997.
[75]B. Russell and D.J. Dix. Am. J. Physiol., 262:C1–C8, 1992.
[76]G.J. Bassell, C.M. Powers, K.L. Taneja, and R.H. Singer. J. Cell. Biol., 126:863–876, 1994.
[77]P. Mahon, J. Beattie, L.A. Glover, and J. Hesketh. FEBS Lett., 373:76–80, 1995.
[78]E.H. Kislauskis, Z. Li, R.H. Singer, and K.L. Taneja. J. Cell. Biol., 123:165–172, 1993.
[79]D. Ferrandon, L. Elphick, C. Nusslein-Volhard, and D. St Johnston. Cell, 79:1221–1232, 1994.
[80]K.M. Forrest and E.R. Gavis. Curr. Biol., 13:1159–1168, 2003.
[81]V.M. Latham Jr., E.H. Kislauskis, R.H. Singer, and A.F. Ross. J. Cell. Biol., 126:1211–1219, 1994.
[82]A.S. Brodsky and P.A. Silver. Methods, 26:151–155, 2002.
[83]B.A. Griffin, S.R. Adams, J. Jones, and R.Y. Tsien. Methods Enzymol., 327:565–578, 2000.
[84]P.J. Santangelo, B. Nix, A. Tsourkas, and G. Bao. Dual FRET molecular beacons for mRNA detection in living cells. Submitted to Nucleic Acids Res., 2004.
[85]S.W. Hell. Nat. Biotechnol., 21:1347–1355, 2003.
HAIRPIN NANOPROBES FOR GENE DETECTION |
435 |
[86]J. Ross. Microbiol. Rev., 59:423–450, 1995.
[87]J. Ross. Trends Genet, 12:171–175, 1996.
[88]N. Heintz, H.L. Sive, and R.G. Roeder. Mol. Cell. Biol., 3:539–550, 1983.
[89]A. Bevilacqua, M.C. Ceriani, S. Capaccioli, and A. Nicolin. J. Cell. Physiol., 195:356–372, 2003.
[90]T.C. Santiago, I.J. Purvis, A.J. Bettany, and A.J. Brown. Nucleic Acids Res., 14:8347–8360, 1986.
[91]Y. Wang, C.L. Liu, J.D. Storey, R.J. Tibshirani, D. Herschlag, and P.O. Brown. Proc. Natl. Acad. Sci. U.S.A., 99:5860–5865, 2002.
[92]S. Pinol–Roma and G. Dreyfuss. Nature, 355:730–732, 1992.
[93]I.E. Gallouzi and J.A. Steitz. Science, 294:1895–1901, 2001.
[94]C.G. Burd and G. Dreyfuss. Science, 265:615–621, 1994.
[95]F. Stutz, A. Bachi, T. Doerks, I.C. Braun, and B. Seraphin et al. Rna, 6:638–650, 2000.
[96]M. Zuker and P. Stiegler. Nucleic Acids Res., 9:133–148, 1981.
[97]J.S. McCaskill. Biopolymers, 29:1105–1119, 1990.
[98]D.J. Ecker, T.A. Vickers, T.W. Bruice, S.M. Freier, and R.D. Jenison et al. Science, 257:958–961, 1992.
[99]D.H. Mathews, M.E. Burkard, S.M. Freier, J.R. Wyatt, and D.H. Turner. Rna, 5:1458–1469, 1999.
[100]M. Andronescu, R. Aguirre-Hernandez, A. Condon, and H.H. Hoos. Nucleic Acids Res., 31:3416–3422, 2003.
[101]Q. Huang and T. Pederson. Nucleic Acids Res., 27:1025–1031, 1999.
[102]J.B. Glotzer, R. Saffrich, M. Glotzer, A. Ephrussi. Curr. Biol., 7:326–337, 1997.
[103]M.R. Jacobson and T. Pederson. Proc. Natl. Acad. Sci. U.S.A., 95:7981–7986, 1998.
[104]M. Somasundaran, M.L. Zapp, L.K. Beattie, L. Pang, and K.S. Byron et al. J. Cell. Biol., 126:1353–1360, 1994.
[105]J.L. Veyrune, G.P. Campbell, J. Wiseman, J.M. Blanchard, and J.E. Hesketh. J. Cell. Sci., 109 ( Pt 6):1185– 1194, 1996.
[106]S. Behrens, B.M. Fuchs, F. Mueller, and R. Amann. Appl. Environ. Microbiol., 69:4935–4941, 2003.
[107]S. Dokka and Y. Rojanasakul. Adv. Drug Deliv. Rev., 44:35–49, 2000.
[108]N.C. Price and L. Stevens. Fundamentals of Enzymology: The Cell and Molecular Biology of Catalytic Proteins. Oxford University Press. New York, 1999.
[109]M.X. Tang, C.T. Redemann, F.C. Szoka Jr. Bioconjug. Chem., 7:703–714, 1996.
[110]O. Zelphati, F.C. Szoka Jr. Proc. Natl. Acad. Sci. U.S.A., 93:11493–11498, 1996.
[111]J.P. Leonetti, N. Mechti, G. Degols, C. Gagnor, B. Lebleu. Proc. Natl. Acad. Sci. U.S.A., 88:2702–2706, 1991.
[112]A.M. Koch, F. Reynolds, M.F. Kircher, H.P. Merkle, R. Weissleder, and L. Josephson. Bioconjug. Chem., 14:1115–1121, 2003.
[113]D.L. Sokol, X. Zhang, P. Lu, and A.M. Gewirtz. Proc. Natl. Acad. Sci. U.S.A., 95:11538–11543, 1998.
[114]C. Molenaar, S.A. Marras, J.C. Slats, J.C. Truffert, and M. Lemaitre et al. Nucleic Acids Res., 29:E89–E89, 2001.
[115]A. Tsuji, H. Koshimoto, Y. Sato, M. Hirano, and Y. Sei-Iida et al. Biophys. J., 78:3260–3274, 2000.
[116]J. Perlette and W. Tan. Anal. Chem., 73:5544–5550, 2001.
[117]D.P. Bratu, B.J. Cha, M.M. Mhlanga, F.R. Kramer, and S. Tyagi. Proc. Natl. Acad. Sci. U.S.A., 100:13308– 13313, 2003.
[118]R.V. Giles, C.J. Ruddell, D.G. Spiller, J.A. Green, and D.M. Tidd. Nucleic Acids Res., 23:954–961, 1995.
[119]M.A. Barry and A. Eastman. Arch. Biochem. Biophys., 300:440–450, 1993.
[120]R.V. Giles, D.G. Spiller, J. Grzybowski, R.E. Clark, P. Nicklin, and D.M. Tidd. Nucleic Acids Res., 26:1567– 1575, 1998.
[121]D.G. Spiller, R.V. Giles, J. Grzybowski, D.M. Tidd, and R.E. Clark. Blood, 91:4738–4746, 1998.
[122]I. Walev, S.C. Bhakdi, F. Hofmann, N. Djonder, and A. Valeva et al. Proc. Natl. Acad. Sci. U.S.A., 98:3185– 3190, 2001.
[123]E.L. Barry, F.A. Gesek, and P.A. Friedman. Biotechniques, 15:1016–1018, 10120, 1993.
[124]S. Paillasson, M. Van De Corput, R.W. Dirks, H.J. Tanke, M. Robert-Nicoud, and X. Ronot. Exp. Cell. Res., 231:226–233, 1997.
[125]B.H. Lloyd, R.V. Giles, D.G. Spiller, J. Grzybowski, D.M. Tidd, and D.R. Sibson. Nucleic Acids Res., 29:3664–3673, 2001.
[126]D. Derossi, G. Chassaing, and A. Prochiantz. Trends Cell. Biol., 8:84–87, 1998.
[127]S.R. Schwarze, K.A. Hruska, and S.F. Dowdy. Trends Cell. Biol., 10:290–295, 2000.