

Micro-Nano Technology for Genomics and Proteomics BioMEMs - Ozkan
.pdf476 |
KIMBERLY HAMAD-SCHIFFERLI |
[51]S.J. Rosenthal, I. Tomlinson, E.M. Adkins, S. Schroeter, S. Adams, L. Swafford, J. McBride, Y. Wang, L.J. DeFelice, and R.D. Blakely. Targeting cell surface receptors with ligand-conjugated nanocrystals. JACS, 124:4586–4594, 2002.
[52]N.C. Seeman and A.M. Belcher. Emulating Biology: Building Nanostructures from the Bottom Up. Proceedings of the National Academy of Sciences, 10:1073, 2002.
[53]W.J. Shaw, J.R. Long, J.L. Dindot, A.A. Campbell, P.S. Stayton, and G.P. Drobny. Determination of statherin N-terminal peptide conformation on hydroxyapatite crystals. JACS, 122:1709–1716, 2000.
[54]R.K. Soong, G.D. Bachand, H.P. Neves, A.G. Olkhovets, H.G. Craighead, and C.D. Montemagno. Powering an inorganic nanodevices with a biomolecular motor. Science, 290:1555–1558, 2000.
[55]M.L. Steigerwald and L.E. Brus. Semiconductor crystallites: a class of large molecules. Acc. Chem. Res., 23:183–188, 1990.
[56]M.N. Stojanovic, T.E. Mitchell, and D. Stefanovic. Deoxyribozyme-based logic gates. JACS, 124:3555–3561, 2002.
[57]A.T. Taton, C.A. Mirkin, and R.L. Letsinger. Scanometric DNA array detection with nanoparticle probes. Science, 289:1757–1760, 2000.
[58]S. Tyagi, and F.R. Krame. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol., 14:303–308, 1996.
[59]S. Wang, N. Mamedova, N.A. Kotov, W. Chen, and J. Studer. Antigen/Antibody immunocomplex from CdTe nanoparticle bioconjugates. Nano Lett., 2:817–822, 2002.
[60]S.R. Whaley, D.S. English, E.L. Hu, P.F. Barbara, and A.M. Belcher. Selection of peptides with semiconductor binding specificity for directed nanocrystal assembly. Nature, 405:665–668, 2000.
[61]E. Winfree, F. Liu, L. Wenzler, and N. Seeman. Design and self-assembly of two-dimensional DNA crystals. Nature, 394:539–544, 1998.
[62]S. Xiao, F. Liu, A.E. Rosen, J.F. Hainfeld, N.C. Seeman, K. Musier-Forsyth, and R.A. Kiehl. Selfassembly of metallic nanoparticle arrays by DNA scaffolding. J. Nanopart. Res., 4:313–317, 2002.
[63]A. Yamazawa, X. Liang, H. Asanuma, and M. Komiyama. Photoregulation of the DNA polymerase reaction by oligonucleotides bearing an azobenzene. Angewandte Chemie-International Edition In English, 39:2356– 2357, 2000.
[64]H. Yang, J.E. Reardon, and P.A. Frey. Synthesis of undecagold cluster molecules as biochemical labeling reagents. 2. Bromoacetyl and maleimido undecagold clusters. Biochemistry, 23:3857–3862, 1984.
[65]M. Yoshida, E. Muneyuki, and T. Hisabori. ATP synthase—a marvellous rotary engine of the cell. Nat. Rev.: Mol. Cell Biol., 2:669–677, 2001.
[66]D. Zanchet, C.M. Micheel, W.J. Parak, D. Gerion, and A.P. Alivisatos. Electrophoretic isolation of discrete Au nanocrystal/DNA conjugates. Nano Lett., 1:32–35, 2001.
[67]S. Zinn and S.L. Semiatin. Elements of Induction Heating, Design Control, and Applications. Metals Park, A.S.M. International, 1988.
16
Sequence Matters: The Influence of Basepair Sequence on DNA-protein Interactions
Yan Mei Wang, Shirley S. Chan and Robert H. Austin
Dept. of Physics, Princeton University, Princeton, NJ 08544, USA
16.1. INTRODUCTION
The sequencing of the human genome, along with the 200-odd other genomes that have been sequenced, does not represent the solution to a puzzle but rather the necessary introduction to a bigger puzzle. That puzzle is how all the 30,000-odd some genes in the human genome are expressed and controlled in a proper sequence for a cell to function. We can hardly address this enormous problem in this brief review, but instead wish to concentrate on one very small but very important aspect of this problem: physical aspects to how proteins are able to achieve base-pair specific recognition. By “physical aspects” we mean that the proteins distort (strain) the DNA helix when they bind, and if this strain is a function of the sequence of the distorted region then the basepair dependent free energy associated with the strain provides a way to discriminate amongst the basepairs.
Our own group has been wrestling with getting DNA containing transcription factors into nanochannels in an attempt to read the proteins coding for genes on a single molecule basis [6, 7, 55, 56]. A major issue that will occur as we shrink structures to the nanometer scale [11] is the deformation of the DNA double helix due to the presence of the transcription factors, and the influence of sequence on the protein binding specificity. We will not address directly the issues of nanotechnology here that are needed to make structures small enough to care about these DNA deformations, but instead hope to lay down some of the basic physical concepts needed for analyzing deformed molecules at the nanoscale.
478 |
YAN MEI WANG, SHIRLEY S. CHAN AND ROBERT H. AUSTIN |
There are three levels of protein-DNA specificity. At a coarse level is the formation of chromatin by the binding of proteins to DNA. While not exquisitely sequence dependent, chromosomes do form very ordered structures and there certainly is a basic sequence dependence to how proteins are guided to form chromosomes during mitosis. At the next level of specificity and complexity are proteins such as restriction enzymes which cut DNA at certain sites. These proteins do not have the on-off control that transcription factors have but they do certainly cut at precision sites with great biological importance. Finally we have the transcription factors, which show the highest specificity of all: binding constants to certain sites approach 10−14 molar, and are 107 times higher than non-specific binding constants to dsDNA.
Consider for example how the λ phage exists within a bacteria. The λ phage is an extremely well known bacteriophage which can operate as a genetic switch: the phage can quietly exist within the genome of its host E. coli (this mode is called the lysogenic phase). However, upon sensing of damage to the genome of the bacterium the phage can switch to a “lytic” phase by exising itself from the host genome and turning off normal control of the phage expression. This removal of the normal control of phage expression results in exponential growth of the phage within the host bacteria causing finally lysis of the bacteria. In order for this switch in expression to occur, there must be a change in the gene expression of phage. In the lysogenic phase a protein called cI binds to a four-fold sequence called OL ,n and OR,n where L or R stands for the left or right side respectively of the cI repressor gene, and n = 1 − 4 for the 4 possible binding sites. Under conditions of lysogeny the expression of cI results in a negative feed-back loop: the cI binds to the OR and OL control sequences. Binding of cI to the OR sequence prevents expression of a protein called cr o to the right and allows for expression of cI to the left side of the operator sequence. Binding of cI to the OL sequences prevents expression to the left side of OL called N. If something happens to the cI repressor protein, the OR and OL repressor sites are no longer occupied with three results: RNA polymerase cannot initiate cI transcription without cI binding to OR , and the proteins cr o and N can now be expressed as the RNA polymerase moves to the right and left respectively of the operator sites. An excellent review of this process can be found in the book “Genetic Switch” by Mark Ptashne [46].
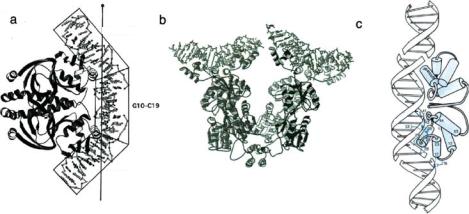
Perhaps the above brief description can give the reader some idea of the amazing amount of highly specific control transcription factors must exert on gene expression in order for an organism to successfully adapt to changing conditions. How does this happen physically, that is, what is the 3-D structure of the protein that allows this to happen? An excellent introduction to this subject can be found in a review article by Luscombe et al. [34]. Generally, transcription factors can be grouped into 4 basic classes, although there are many exceptions to the following list: (1) helix-turn-helix, (2) zinc finger, (3) leucine, (4) helix-loop-helix. For all 4 cases the basic structure consists of two DNA binding sections which are separated by a linker region. The linker region does not contact the DNA helix. Why is this particular structure so predominantly chosen? We can guess that perhaps part of the recognition process involves some sort of straining of the helix in the non-contacted region. Figure 16.1 shows some examples of how proteins can deform the double helix upon binding.
This bending of the helix which would make some sense as a means of enhancing specific if in fact the elastic properties of DNA are a function of the basepair composition, for then the strain energy would stored in the strained helix would vary as a function basepair sequence in the strained sequence. In order to test this supposition three subjects must be
SEQUENCE MATTERS: THE INFLUENCE OF BASEPAIR SEQUENCE |
479 |
a |
A20–G29 |
|
b |
|
c |
||
|
|
||||||
|
|
|
|
|
|
||
|
|
|
|
63 |
|||
|
|
|
|
|
|
||
|
|
G10–C19 |
|
||||
|
|
|
7 |
||||
6 |
|
|
|
|
5 |
|
|
|
|
4 |
|
|
|
|
3 |
|
|
|
|
2 |
|
|
|
|
|
|
28 |
|
|
|
1 |
|
|
|
|
|
|
||
|
16 |
|||
G1–T9 |
|
|
|
|
FIGURE 16.1. Dimer binding of (a) CAP, (b) LacR protein and (c) phage 434 repressor protein to DNA. The bending angles are 90◦ in CAP, 42◦ in LacR protein and 25◦ in phage 434 repressor protein. LacR protein in nature binds as tetramers to two isolated DNA. LacR protein and phage 434 repressor protein bind to DNA with helix-turn-helix motif. Adapted from Refs. [17] [31] and [1].
addressed: (1) we have to have a mathematical formalism for the strain energy stored in a deformed helix; (2) we have to have measurements of the elastic constants of the helix as a function of basepair sequence; (3) we need to bring the previous parts together and show that experiments support the supposition that sequence-dependent elastic properties of DNA and strain can explain a significant part of the free energy of sequence dependent specificity in transcription factors.
The structure of Deoxyribose Nucleic Acid (DNA) was first proposed by Watson and Crick in 1953 [57]. In their model, DNA is a double helix of anti-parallel sugar-phosphate chains held together by a stack of complementary base-pairs (bp). Base-pairs contain four base: adenine (A) and guanine (G) (purine), cytosine (C) and thymine(T) (pyrimidine). Purine bases always pair with pyrimidine bases; i.e., A pairs with T (A·T) and G with C (G·C). Purine and pyrimidine in a base pair are connected by hydrogen bonds—two for A·T and three for G·C. This DNA is called a classical Watson-Crick B-form DNA. In real life, the Watson-Crick DNA of perfect helix form with all its bases, sugars and phosphates at their assigned locations, rarely exists. What exists is a double helix with local (of order base-pair separation distance) structural variations brought about by generic differences of the stacking four bases. Since each base has its own unique composition and thus electronic distribution, it is conceivable that for different base sequences, equilibrium separation and rotation between bases would be different. For example, it would not be hard to imagine that DNA homo-polymer such as polyd(G·C) where the long purine bases reside on one chain having different conformation from alternating DNA polymer such as polyd(GC).
It is now generally accepted that properties of DNA (structural, mechanical etc.) depend on base-pair sequence. While sugar phosphate backbone ensures the continuous double helical form of DNA, it is mainly the sequence of bases that determines DNA local geometrical properties. In order to discuss the sequence’s effects on DNA, it is necessary to first define all relevant base movements in DNA. Translational and rotational parameters for relative displacements of base to base or base-pair to base-pair were established and standardized in 1989 [16, 18]. Figure 16.2 shows all coordinate parameters alone which translations


SEQUENCE MATTERS: THE INFLUENCE OF BASEPAIR SEQUENCE |
481 |
and rotations of bases and base-pairs are performed. Among these translational and rotational parameters, twist (propeller twist and helical twist), roll, tilt and slide are sufficient in describing sequence dependent DNA properties and DNA-protein interactions in this article.
16.2. GENERALIZED DEFORMATIONS OF OBJECTS
We relied heavily in this section on Landau and Lifshitz Vol. 7 (Elasticity) [32] and an informative article by Goldstein, Powers and Wiggins [22]. We first note that it is possible to write the displacement δr of vector r when it is rotated through an angle δφ
as a cross-product of the vector |
|
|
|
|
|
and r : |
|
|
|
||
|
δφ |
|
|
|
|
|
|
= |
δφ |
× |
(16.1) |
|
|
δr |
|
r |
|
which you can convince yourself is true by taking the vector at some finite angle φ to the x axis and rotating a bit through the angle δφ. Now, imagine that an orthonormal coordinate system ei is attached to the filament at some point. Let e1 and e2 be the x and y axis respectively in the cross-sectional plane of the filament and let e3 be the z axis parallel to the local tangent, ie, e3 is the tangent vector. The deformation of the filament is the
local rotation d of this coordinate system as we move an arc length element ds along the
φ
filament. It is common to use the vector to represent this deformation:
d
= φ (16.2)
ds
The strains of the object are then proportional to the rate at which the coordinate system rotates:
|
|
|
∂ei |
|
|
e |
(16.3) |
|
|
|
|
||||
|
|
|
∂s |
= |
|
i |
|
|
|
|
|
× |
|
||
Those elements of |
|
|
|
|
|
|
|
which are parallel to the e3 axis are defined as torsional deformations, |
|||||||
|
|
|
|
|
|
|
|
while those elements along e1, e1 are bending deformations.
We can make this more explicit. Note that de3/ds is the curvature of the filament, and
is equal to 1/R, where R is the radius of curvature of the filament. Hence: |
|
||||||
|
de3 |
|
|
e |
3 |
n |
(16.4) |
|
ds |
= |
|
|
= R |
|
|
|
|
× |
|
||||
where n is a unit vector in the plane of curvature and perpendicular to the tangent e3:
n |
R |
de3 |
(16.5) |
|
ds |
||||
= |
|
|
If we cross the tangent vector e3 with both sides of Eq. 16.4 and use the vector identity:
A × (B × C) = B( A • C) − C( A • B) |
(16.6) |

SEQUENCE MATTERS: THE INFLUENCE OF BASEPAIR SEQUENCE |
483 |
Integration over the entire cross-section yields the total restoring torque τ :
τ = |
2π Gα |
|
r 3dr = |
Gα |
IP |
(16.12) |
L |
L |
where IP is defined as the polar moment of inertia:
IP = 2π r 3dr (16.13)
Note that IP varies as the fourth power of the radius of the rod, as does the surface moment of inertia IA. We now see that since α/L is the rotational rate 3 of the coordinate system and that the twisting rigidity C can be written in general as:
C = |
G Ip |
(16.14) |
L |
A final note. There is a connection between the thermally induced bending and twist-
ing dynamics of the DNA molecule and the elastic modulii. Thermal energy stores kB T
2
of energy per degree of freedom in a system, where kB is Boltzmann’s constant and T is the temperature in Kelvin. This thermal energy results in a bending and twisting persistence length pB and pT respectively [32]. These lengths are basically the average radius of curvature due to thermal induced bending and the average distance over which the helix twists through an RMS angle < φ2 >1/2 2π . The bending persistence length is:
E Ia |
(16.15) |
pB = kB T |
while the twisting persistence length is:
G Ip |
(16.16) |
pT = kB T |
The bending persistence length is quite easy to observe since it results in the deformation of the backbone of the dsDNA molecule giving rise to a random walk aspect to the contour of the molecule which can be directly measured as the radius of gyration Rg of the polymer. Rg is basically the radius of the glowing blob that a genomic length dsDNA molecule appears as in a microscope. For a non-self avoiding random walk Rg is given for a polymer of contour length L and bending persistence length pB by [19]:
Rg = |
L6 B |
1/2 |
||
(16.17) |
||||
|
|
p |
|
|

484 YAN MEI WANG, SHIRLEY S. CHAN AND ROBERT H. AUSTIN
TABLE 16.1. Elastic properties of defined length DNA sequences
Sequence |
E (dyne cm−2) |
pB (bp) |
poly(dG) poly(dC) |
2.3 ×109 |
400 |
poly(dA-dC) poly(dT-dG) |
1.4 ×109 |
250 |
poly(dA) poly(dT) |
8.2 ×108 |
150 |
16.3. SEQUENCE DEPENDENT ASPECTS TO THE DOUBLE HELIX ELASTIC CONSTANTS
The next question is if there is a DNA sequence dependence to E and G. This sequence dependence could come from at least two interactions: “on diagonal” terms due to the differences in the hydrogen bonding patterns of G· C and A · T basepairing (denoted by the symbol ·), and “off-diagonal” terms due to differences between the basepair stacking interactions of nearest neighbor bases, such as G-C, denoted by the hyphen. The effect of sequence on DNA elastic properties (torsional and bending stiffness) was investigated in our group with a few hundred bp long DNA segments using triplet anisotropy decay techniques [25]. In this work DNA was treated as a stack of base-pairs connected by springs. Table 16. compares E for different three sequences: poly(dG)·poly(dC), poly(dA-dC)·poly(dT-dG), poly(dA)·poly(dT) and the predicted bending persistence length pB as calculated from Eq. 16.15. These measurements showed that AT rich sequences where the most flexible and GC rich sequences where the least flexible.
Other experimental techniques have been used in studying DNA flexibility: they are scanning force microscopy [49], nuclear magnetic resonance [38], fluorescence polarization anisotropy (FPA) measurements [21] and electron paramagnetic resonance [40]. There is by no means an agreement in the literature that the values we cite in Table 16.1 are gospel truth. Hagerman in his 1988 review article [23] dismissed the results shown above as “probably artifactual”. But, we will see that this story has continued in spite of his sceptism. There are probably strong basepair dependences to DNA physical properties, as a function of sequence [5], that is no doubt more complex than the simple picture presented above, but to deny the existence of any substantial basepair dependence to the elastic properties of DNA is surely wrong.
That there are substantial sequence dependences is clear from other areas of research. There is a thermodynamic literature which connects melting points of DNA oligonucleotides and basepair composition, a more indirect approach than the physical approaches discussed above but indicative of the substantial dependence of basepair composition on the physical properties of DNA. Thermodynamic measurements which measure the heat exchange Q when molecules interact basically yield the values for the enthalpy changes H . Enthalpy is not the same as the potential energy U . Enthalpy is one of the possible “free energy” potential measurements of a system. The free energy of a system is the extactable energy you can remove from a system subject not only to the constraints of the conservation of energy E, but also subject to the constraint that a physical system will always want to be in the macrostate of maximum entropy S, it wants to be as disordered as it can be. Thus, enthalpy changes H are more complex quantities than the simpler elastic potential energy

SEQUENCE MATTERS: THE INFLUENCE OF BASEPAIR SEQUENCE |
485 |
TABLE 16.2. Nearest Neighbor Thermodynamics
Interaction |
H ◦ cal/M |
S◦ cal/K-M |
G◦ cal/M |
AA/TT |
9.1 |
24.0 |
1.9 |
AT/TA |
8.6 |
23.9 |
1.5 |
TA/TA |
6.0 |
16.9 |
0.9 |
CA/GT |
5.8 |
12.9 |
1.9 |
GT/CA |
6.5 |
17.3 |
1.3 |
CT/GA |
7.8 |
20.8 |
1.6 |
GA/CT |
5.6 |
13.5 |
1.6 |
CG/GC |
11.9 |
27.8 |
3.6 |
GC/CG |
11.1 |
26.7 |
3.1 |
GG/CC |
11.0 |
26.6 |
3.1 |
|
|
|
|
changes we discussed earlier but do contain within them some (unknown) elements of the total thermodynamic differences between basepairs in DNA.
Breslauer’s group at Rutgers University has been the leader in the field of measuring basepair dependent thermodynamic properties of DNA, predominantly in basepair specific specific heat changes [4, 54]. By analyzing the basepair dependent melting temperatures of oligonucleotides it has been possible to construct a table of basepair-basepair stacking interactions which include as we have mentioned both the elastic potential energy effects we discussed above and entropic issues which we have ignored. Table 16.2 is extracted from [4] and gives in tabular form the thermodynamic variances in stacking interactions. As the authors state in [4], “base sequence and not base composition determines stability”. Note also that the sequence AA/TT, what we will call an “A-tract” in the next section, has an anomalous stability. The subtle consequences of A-tracts will be clarified next, but the point here is that the Gibbs free energy G◦ varies by factors of 3 with basepair sequence, hardly a small effect.
16.4. SEQUENCE DEPENDENT BENDING OF THE DOUBLE HELIX AND THE STRUCTURE ATLAS OF DNA
There is more to the picture of the conformational complexity of DNA than just the variation of the elastic constants with basepair sequence as is clear from the thermodynamic results. DNA is held together with hydrogen bonds, and these bonds can connect atoms not only directly across the basepairs but also off-diagonally to neighboring groups. In order to maximize the energy of these bonds, the basepairs typically twist out of the plane and can form what is called a bifurcated hydrogen bond. Not all bases can do this, the AA-TT sequence has the least amount of steric clash and so can do this most readily if the As are all on one side and the Ts are all on the other: this is called a homo-A tract. The designation of a homo A-tract is poly (dA)·poly (dT). So, strangely, although we would expect that a random sequence of A·T basepairs to be the most flexible, this rule breaks down for A tracts because of the cross-coupling of the bifurcated hydrogen bond which can enhance the rigidity of the helix, at least at temperatures below 30 C. The formation of the bifurcated hydrogen bond results in a propellor twist to the bairpairs as they rotate out of the plane