

Реакция с радикалами.
Этот вопрос изучен мало; известно лишь, что в пиримидине С5-атом наименее чувствителен к атаке арильных радикалов, а в пиридазине четвертое положение, вероятно, обладает наиболее высокой реакционной способностью. В случае взаимодействия пиразина и хинолина с радикалами кольцевые атомы азота являются главными участками атаки[10,11].
Металлирование галогендиазинов
В 5-бромпиримидинах замещение галогена атомом металла происходит при очень низкой температуре. При более высоких температурах (—80°С) реакция замещения сопровождается присоединением[11].
Схема 25

Реакционноспособность заместителей боковой цепи
Метилпиразин реагирует с амидом натрия в жидком аммиаке, образует анион, который может быть алкилирован, образуя алкилпиразины (схема 26).
Схема 26
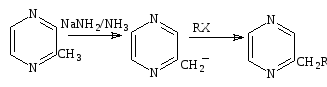
Алкилпиразины нашли широкое употребление в качестве ароматических веществ. Распространены реакции с альдегидами и кетонами, например, метилбензоат приводит к образованию фенацилпиразина с 95% выходом.
Схема 27

2 – Метилхиноксалин реагирует с формальдегидом, образуя22и23с низким выходом.

22 23
2,3 – диметилхиноксалин реагирут подобным образом.
Алкилпиразины и алкилхиноксалины участвуют в реакции Манниха cформальдегидом и диметиламином, образуя диалкиламиноэтилпроизводные24и25, которые служат исходным материалом для получения соответствующих виниловых производных[12,13].

24 25
Образование аниона было достигнуто при использовании очень сильных оснований, таких как BuLi (Схема 28). Сложность данной реакции в том, что необходимы металлоорганические реактивы, не затрагивающиеC=N. Данные реакции приводят к образованию 1,2 – дигидропиразинов, которые подвергаются окислению.
Схема 28

Схема 29

Так же сложность при использовании сильных оснований – это возможность образования аниона в незамещенном кольцевом положении, смежном с атомом азота 26.
Алкилированные боковые цепи в пиразине и хиноксалине восприимчивы к галогенам, и в радикальных условиях к NBS. Таким образом, бромирование 2 – метилксиноксалина с с бромом в присутствии ацетата натрия дает выход только трибромметилхиноксалина27.
Хлорирование метилпиразина в уксусной кислоте при 100°С дает тоже трихлорметилпиразин 28. Но когда идет хлорирование 2 – хлор – 3 – метилпиразина образуется 2- хлор – 3 – дихлорметилпиразин29.
26 27 28 29
Бромирование 2 – этил – 3 – метилпиразина дает 2 – бромэтил – 3 – метилпиразин с количественным выходом, он может быть окислен солью натрия 2 – нитропропана или пиридином в окиси, чтобы получить 2 – ацетил – 3 – метилпиразин с выходом 66% и 25% соответственно (схема 30)[14,46].
Схема 30
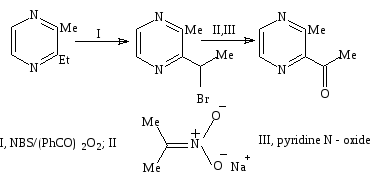
Окисление
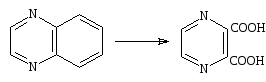
N – оксиды. К действию окислителей пиразин менее устойчив, чем пиридин, и обесцвечивает щелочной раствор перманганата на холоду. Пиразинкарбоновые кислоты хотя и могут быть получены окислением алкильных боковых цепей, но выходы при этом обычно бывают ничтожны. В хиноксалинах пиразиновый цикл несколько более устойчив, чем бензольный, и превращение хиноксалина в пиразиндикарбоновую-2,3 кислоту происходит быстро и с хорошим выходом[15].
Схема 31
Алкилпиразины реагируют с перекисью водорода, давая смесь моно- и ди-N-окисей.
Схема 32
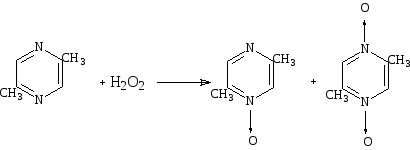
Действием перекиси водорода в уксусной кислоте на диазины получены их моно-N-окиси; как и можно было предвидеть, образование ди-N-окисей легче всего идет с пиразинами, тогда как в случае пиридазина выход составляет ~ 1 %.
Схема 33

Окиси гетероциклов занимают ключевую позицию в химии гетероциклов, они объясняют функциональную подвижность группы и структурные возможности модификации, не доступные любым другим соединениям.
Обычно проводят прямое окисление, используя H2O2/AcOH, хотя и существует опасность взрыва. Окиси пиразина30, хиноксалина31и феназина32получают с высоким выходом[16,17].
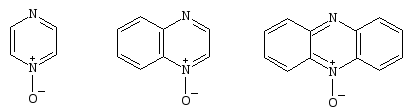
303132
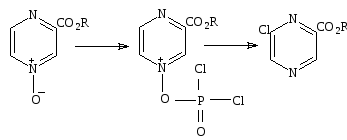
Обработка окиси пиразина и окиси хинолина РОCl3приводит к образованию хлорпроизводных33, 34.

33 34 35 36
Однако в случае 1 - окиси 2- хлорхиноксалина образуется 6 – хлорохиноксалин (35) .
1,4 – диоксид 2,3 – диметилпиразина реагирует с уксусным ангидридом, образуя 2,3 – ацетоксиметилпиразин (36). Обработка 3 - окиси – алкоксикарбонилпиразина оксохлоридом фосфора не приводит к образованию ни одного из ожидаемых продуктов, содержащих хлор, образуется 2 – хлоро – 6 – метоксикарбонилпиразин[18].
Схема 34
Кольцевые заместители показывают высокую реакционноспособность к нуклеофильному замещению, относительно неокисленных систем в 3 положение. В случае хиноксалина и феназина степень активности данного заместителя, зависит от того стабилизирован ли промежуточный комплекс за счет мезомерного эффекта. 2 – хлорпиразин 1 - оксид с легкостью переходит в 2 – гидроксипиразин 1 – оксид 37при обработке водным гидроксидом натрия, тогда как 2,3 – дихлорпиразин и 3 – хлорпиразин 1 – оксиды устойчивы в этих условиях[47,48].

37
В случае N– оксидов феназина, наблюдается активность к нуклеофильному замещению. 1 – хлорфеназин является обычно очень стойким к нуклеофильному замещению, но его второй изомер более реакционноспособный и возможно замещение различными нуклеофильными агентами[19,20].
Размыкание цикла.
Хотя пиразины, в общем, устойчивы к щелочам и кислотам, опыты с некоторыми арилпроизводными показывают, что йодистоводородная кислота может расщепить пиразиновое ядро. По-видимому, этот реагент сначала восстанавливает пиразин до дигидропроизводного, которое легко гидролизуется. Тип получаемых продуктов реакции зависит от положения заместителей. 2,5-Дифенилпиразин дает ω-аминоацетофенон, а 2,6-дифенилпиразин—дифенациламин. Механизмы этих реакций небыли изучены, но следующий ряд превращений был бы возможен[19,20,21]:
Схема 35
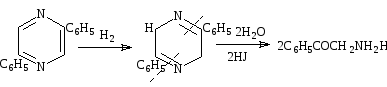
Схема 36

![]()