

2.4 Получение пиразинов
Классический метод получения пиразинов, т. е. конденсация α-аминокарбонильных соединений самих на себя, малопригоден для синтеза родоначального члена ряда. Необходимое для этого аминокарбонильное соединение—α-аминоацетальдегид—склонно давать различные побочные реакции, и по этой причине аминирование бромацетальдегида [29] и гидролиз аминоацеталя [8, 30] приводят лишь к очень низким выходам пиразина.
Схема 3

Пиразин был получен также обходным путем — декарбоксилированием пира-зинкарбоновых кислот [7, 29]. Однако в течение многих лет самым продуктивным методом синтеза пиразина был метод Вольфа и Марбурга [28] из ацеталя иминодиацетальдегида. При обработке последнего соединения соляной кислотой ацетальные связи разрываются и образуется 2,6-диоксиморфолин. Это производное морфолина реагирует с гидроксиламином, давая пиразин:
Схема 4

Пиразин получается с 78%-ным выходом непосредственно из ацеталя при обработке последнего солянокислым гидроксиламином. Главный недостаток этого метода синтеза заключается в том, что исходное вещество представляет собой труднодоступное соединение.
Более современные патенты были приняты для промышленных методов получения пиразина. Это продолжительные процессы, включающие высокотемпературные реакции в паровой фазе над подходящими катализаторами. Пиперазин может быть дегидрирован до пиразина с удовлетворительными выходами над оксидами некоторых тяжелых металлов или дегидрирующими катализаторами, такими, как хромит меди или палладий . Так как пиперазин получается дегидратацией м-(2-оксиэтил)-этилендиамина в сходных условиях, два процесса могут быть объединены в одну стадию. При пропускании паров оксиэтилэтилендиамина или диэтилентриамина над смешанным дегидратирующим и дегидрирующим катализатором (активированный оксид алюминия, покрытый никелем) при 400° получается непосредственно пиразин с общим выходом около 50% [26].
Схема 5

На ранних этапах главное внимание в химии пиразина было уделено получению и изучению пиразинов с идентичными углеводородными заместителями в 2,5- и 2,3,5,6-положениях, так как эти соединения составляют наиболее легко доступный класс производных. а-Аминокарбонильные соединения, будучи в растворе в свободном состоянии, самопроизвольно конденсируются в дигидропиразины, которые затем окисляются до соответствующих пиразинов. К числу обычных окислителей относятся хлорная ртуть [8,30], перекись водорода [19, 28] и воздух. Часто превращение дигидропиразина в пиразин происходит настолько легко, что промежуточное дигидросоединение не может быть выделено.
Одним из наиболее широко применяемых методов получения необходимых α-аминокетонов является восстановление соответствующих оксиминокетонов, которые в свою очередь получаются нитрозированием кетонов. Старая методика восстановления, использующая хлористое олово в соляной кислоте, дает аминокетон в виде соли. В процессе выделения основания действием щелочи образуется дигидропиразин. Другими обычными восстановителями оксимино кетонов в пиразины являются цинк и щелочь, цинк и уксусная кислота и амальгама алюминия. Электролитическое восстановление оксимино-ацетона дает лишь ничтожные выходы 2,5-диметилпиразина [31]. Попытки получить а-аминоспирты каталитическим восстановлением соответствующих а-оксиминокетонов иногда приводят к образованию дигидропиразинов [32, 33]. Промежуточным продуктом по крайней мере некоторых из этих реакций может быть скорее иминоспирт, чем аминокетон [34], так как каталитическое гидрирование бензоиноксима также дает небольшие количества производных пиразина [32]. Восстановление диоксимов приводит в некоторых случаях к производным пиразина [31, 32, 33], которые образуются, по-видимому, через оксиминокетоны.
Схема 6

Простые монооксимы могут быть превращены в α-аминокетоны или пиразины путем необычной перегруппировки их сложных эфиров с п-толуолсульфокислотой [34], которая происходит под действием этилата калия.
Схема 7

При этом можно либо выделить соль аминокетона, либо получить непосредственно пиразин или дигидропиразин. Тщательное изучение механизма показывает, что в этой реакции образуются промежуточные циклические соединения, такие, как нестойкие производные этоксиэтиленимина 5.
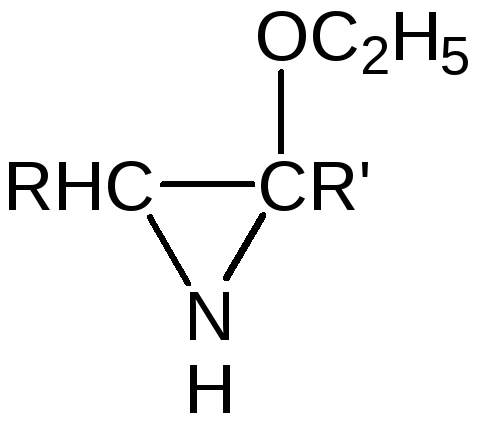
5
Хотя симметричные пиразины были получены непосредственным взаимодействием α-галогенированных карбонильных соединений с аммиаком [34, 35— 37], применимость этого метода сильно ограничивается различными побочными реакциями. Более четкий фталимидный метод синтеза аминов в общем случае применим и для получения аминокетонов и является поэтому предпочтительным [8, 34].
Аминокислоты превращали в α-аминокарбонильные соединения двумя различными методами. Неуберг восстанавливал сложные эфиры глицина и аланина амальгамой натрия и получил с низкими выходами пиразин и 2,5-диметилпиразин соответственно. В более практическом методе N-ацильные производные а-аминокетонов получаются нагреванием а-аминокислот с ангидридами или фторангидридами кислот в присутствии пиридина [38]. При гидролизе эти соединения дают аминокетоны. Одним из промежуточных продуктов может быть азлактон (XII), который ацилируется в реакционноспособное положение 4, а затем разлагается с выделением двуокиси углерода и образованием ациламинокетона.
В отдельных случаях 2,5-дизамещенные и 2,3,5, 6-тетразамещенные пира-зины были получены не только из α-аминокетонов, но и из других соединений. Перегонка глицерина со смесью хлорида и фосфата аммония дает смесь оснований, содержащую немного 2,5-диметилпиразина и небольшое количество 2,5-диметил-3-этилпиразина [39]. Этот синтез включает, по-видимому, промежуточное образование акролеина. Конденсация двух молекул последнего с одной молекулой аммиака дает β-метилпиридин, тогда как взаимодействие акролеина и аммиака (по две молекулы каждого) приводит к образованию диметилпиразина 7.
Схема 9

6
Схема 10

7
Следовательно, большой избыток применяющегося аммиака способствует образованию пиразина. Добавление к реакционной смеси ацетальдегида повышает выход 2,5-диметил-З-этилпиразина; по-видимому, ацетальдегид может быть промежуточным продуктом в образовании этого производного.
Некоторые оксикарбонильные соединения превращаются прямо в пиразины обработкой аммиаком или солями аммония. Хорошие выходы тетрафенилпиразина 8были получены при нагревании бензоина с аммиаком, муравьинокислым или уксуснокислым аммонием.
Схема 11

8
2,3-дизамещенные пиразины могут быть получены реакцией 1,2-диаминовс 1,2-дикетонами с последующей дегидрогенизацией образовавшихся дигидропроизводных при нагревании [42-44]. Если заместители ароматические, то конденсация происходит гладко и с хорошим выходом.'В случае применения вышеуказанной реакции к С-замещенным эти-лендиаминам [40] могут быть получены некоторые три- и тетразамещенные пиразины.
Схема 12

Схема 13

Триметилпиразин получается с очень низким выходом вместе с тетраметилпи-разином при нагревании бромметилата 2,5-диметилпиразина до 270—280° [41]. Попытки металлировать метильную группу в 2,5-диметилпиразине привели к открытию реакции, позволяющей вводить еще один заместитель в ме-тилпиразины. При обработке 2,5-диметилпиразина этил- или фениллитием ожидаемые литиевые производные не образуются, а вместо этого происходит присоединение по азометиновой связи с образованием тризамещенного пиразина [39].
Схема 14

С самим пиразином реакция протекает с ничтожным выходом.