

Reference_01_08_2014_165529
.pdfSciFinder® |
Page 1 |
1. Enzymatic resolution by CALB of organofluorine compounds under conventional condition and microwave irradiation
By Ribeiro, Sandra S.; Raminelli, Cristiano; Porto, Andre L. M.
From Journal of Fluorine Chemistry (2013), 154, 53-59. Language: English, Database: CAPLUS, DOI:10.1016/j.jfluchem.2013.06.014
Enzymic kinetic resoln. of organofluorine rac-alcs. by CALB yielded (-)-(R)-2,2,2-trifluoro-1-phenylethanol, (-)-(R)-1-(3- bromophenyl)-2,2,2-trifluoroethanol, (-)-(R)-1-(4-bromophenyl)-2,2,2-trifluoroethanol, (-)-(S)-1-(2,4,5- trifluorophenyl)ethanol, (+)-(S)-2,2,2-trifluoro-1-phenylethyl acetate, (+)-(S)-1-(3-bromophenyl)-2,2,2-trifluoroethyl acetate, (+)-(S)-1-(4-bromophenyl)-2,2,2-trifluoroethyl acetate and (+)-(R)-1-(2,4,5-trifluorophenyl)ethyl acetate in high enantiomeric excess (up to >99% ee). The reactions were conducted under conventional conditions (orbital shaking) and microwave irradn. in toluene and vinyl acetate as acylating agent. The CALB showed excellent selectivities and good yields in the transesterification of fluorinated arom. compds.
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
2. Highly efficient synthesis of aryl and heteroaryl trifluoromethyl ketones via o-iodoxybenzoic acid (IBX)
By Cheng, Huicheng; Pei, Yu; Leng, Faqiang; Li, Jingya; Liang, Apeng; Zou, Dapeng; Wu, Yangjie; Wu, Yusheng From Tetrahedron Letters (2013), 54(33), 4483-4486. Language: English, Database: CAPLUS, DOI:10.1016/j.tetlet.2013.06.045
A 2-step process for the synthesis of aryl and heteroaryl trifluoromethyl ketones from the corresponding aldehydes is described. Trifluoromethyl alcs. were prepd. from aryl and heteroaryl aldehydes in excellent yields using catalytic amts. of K2CO3. The trifluoromethyl alcs. were then oxidized conveniently and efficiently by 2-iodoxybenzoic acid (IBX) under mild conditions to give the desired functionalized aryl and heteroaryl trifluoromethyl ketones.
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
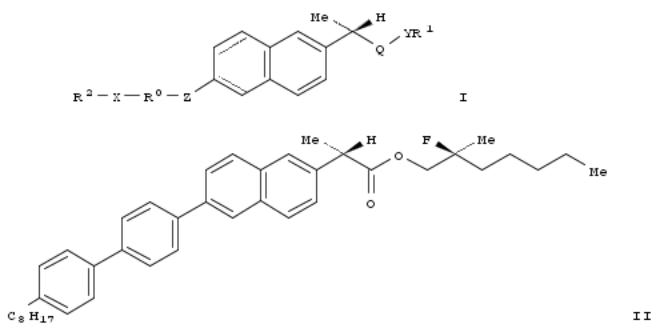
3. Naproxen based ferroelectric liquid crystal or nematic liquid crystal chiral compounds useful for electro-optical and display device applications and their preparation
By Zhang, Yongqiang; More, Kundalika; Thurmes, William
From U.S. Pat. Appl. Publ. (2013), US 20130172558 A1 20130704, Language: English, Database: CAPLUS
The invention is related to naproxen based ferroelec. liq. crystal (FLC) or nematic liq. crystal chiral compds. of formula I, which can be used for electro-optical and display device applications and their prepn. The invention compds. I, wherein R0 is arom. ring system, cyclic ring system, hetero arom. ring system, etc.;
R1 and R2 are C2-20alkyl, C2-20alkenyl, or C2-20alkynyl; Q is CH2, CO, or CS; X is O, OCO, OCH2, etc.; Y is O or S; are
claimed. Compd. II was prepd. by multi-step procedure (procedure given). Also provided is a method of prepg. a liq. crystal display device on silicon that includes incorporating I into a liq. crystal display on silicon by disposing the compd. in a liq. crystal, or the liq. crystal, onto a silicon surface.
SciFinder® |
Page 2 |
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
4. Solvent selection in synthesis of 4-(1-arylfluoroethoxy)quinazolines and thienopyrimidines
By Han, Jin; Sundby, Eirik; Hoff, Baard Helge
From Journal of Fluorine Chemistry (2013), 153, 82-88. Language: English, Database: CAPLUS, DOI:10.1016/j.jfluchem.2013.05.011
The nucleophilic arom. substitution of 4-chloroquinazoline and 6-bromo-4-chlorothieno[2,3-d]pyrimidine with 1-aryl-2- fluoroethanols as nucleophiles has been studied focusing on the use of carbonate bases in combination with environmentally acceptable solvents. The conversion rate depended on the solvent properties, the acidity of the nucleophile and the nature of the base. By using acetonitrile as reaction medium and K2CO3 as base, 2,2,2-trifluoro-, 2,2-difluoro-, and 2-fluoro-1-phenylethanol could efficiently be coupled to 4-chloropyrimidines. Alternatively, employing Cs2CO3, allowed for shorter reaction time for these substrates, and also couplings of the nonfluorinated alcs. proceeded well. Tert-butanol was also found to be a suitable reaction medium in transformation of the fluoro alcs. Testing of hydrolytic stability of the 4-alkoxypyrimidines revealed that the fluorinated and nonfluorinated derivs. were labile under acidic conditions, whereas in basic media the fluoroalkoxy derivs. were more stable than their nonfluorinated counterparts.
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
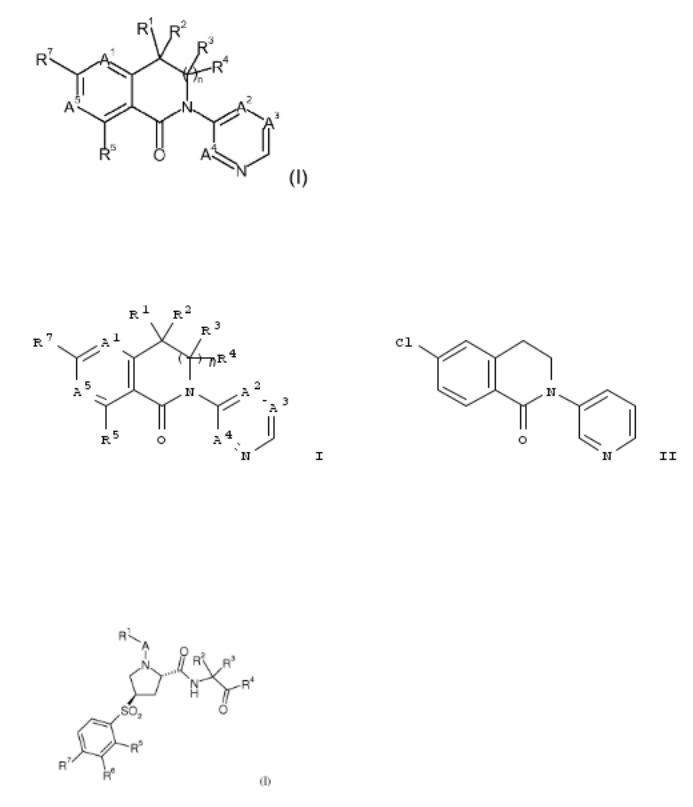
5. New bicyclic dihydroisoquinolin-1-one derivatives as aldosterone synthase inhibitors and their preparation
By Aebi, Johannes; Amrein, Kurt; Chen, Wenming; Hornsperger, Benoit; Kuhn, Bernd; Liu, Yongfu; Maerki, Hans P.; Mayweg, Alexander V.; Mohr, Peter; Tan, Xuefei; et al
From PCT Int. Appl. (2013), WO 2013079452 A1 20130606, Language: English, Database: CAPLUS
SciFinder® |
Page 3 |
The invention provides compds. having formula I, compns. including the compds. and methods of using the compds. as aldosterone synthase (CYP11B2 or CYP11B1) inhibitors for the treatment or prophylaxis of chronic kidney disease, congestive heart failure, hypertension, primary aldosteronism and Cushing syndrome. Compds. of formula I wherein E is absent and CR3R4; A1 is CR8 and N; A2 is CR9 and N; A3 is CR10 and N; A4 is CR11 and N; A5 is CR6 and N; R1, R2, R3 and R4 are independently H, alkyl, cycloalkyl, haloalkyl, etc.; R2R4 can be taken together to form a double bond, then R5 is H; R1R2, R3R4 or R1R3 can be taken together to form substituted cycloalkyl and heterocycloalkyl; R5, R6, R7 and R8 are independently halo, CN, alkoxy, hydroxyalkoxy, etc.; R9 is H, halo, OH, CN, alkyl, etc.; R10 is (un)substituted alkoxy and substituted alkyl; R11 is H; and with proviso; and pharmaceutically acceptable salts thereof, are claimed. Example compd. II was prepd. by N- arylation of 6-chloro-3,4-dihydroisoquinolin-1(2H)-one with 3- bromopyridine. The invention compds. were evaluated for their aldosterone synthase inhibitory activity. From the assay, it was detd. that compd. II exhibited EC50 values of 0.0860 μM and 4.6072 μM towards CYP11B2 and CYP11B1, resp.
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
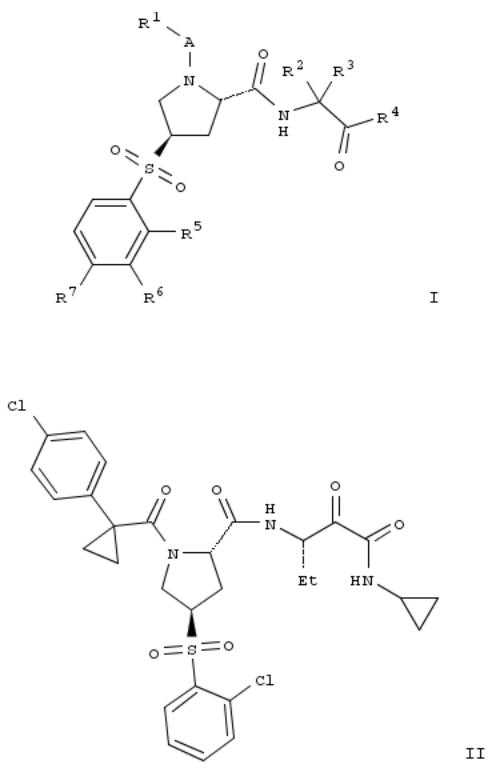
6. Preparation of substituted 4-(phenylsulfonyl)pyrrolidine-2-carboxamide derivatives as cathepsin inhibitors
By Banner, David; Haap, Wolfgang; Kuhn, Bernd; Luebbers, Thomas; Peters, Jens-Uwe; Schulz-Gasch, Tanja From PCT Int. Appl. (2013), WO 2013076063 A1 20130530, Language: English, Database: CAPLUS
Title compds. I [A = carbonyl or absent; R1 = alkoxy, nitrophenyl, alkylcycloalkyl, haloalkylcycloalkyl, phenylcycloalkyl, etc.; R2 and R3 = independently H, alkyl, cycloalkylalkyl or cycloalkyl when taken together; R4 = C(O)NR8R9 or benzooxazolyl; R5, R6 and R7 = independently H, alkyl, halogen, haloalkyl, alkoxy, etc.; one of R8 and R9 = H or alkyl, and the other one = alkyl, alkoxyalkyl, cycloalkyl, haloalkyl, phenylalkyl, etc.], and their pharmaceutically acceptable salts, are prepd. as cathepsin inhibitors. Compd. II was prepd. by coupling of (2S,4R)-1-[1-(4- chlorophenyl)cyclopropanecarbonyl]-4-(2- chlorophenylsulfonyl)pyrrolidine-2-carboxylic acid (prepn. given) with (2S,3S)-3-amino-N-cyclopropyl-2- hydroxypentanamide dihydrochloride followed by oxidn. of the hydroxyl group. Compd. II exhibited inhibitory activity against human cathepsin S, L, K and B with IC50 values of 0.000296 μM, 0.0043 μM, 0.0276 μM and 0.0627 μM, resp. in the cathepsin enzyme inhibition assay. The invention compds. are useful for the treatment of diabetes, atherosclerosis, abdominal aortic aneurysm, etc.
SciFinder® |
Page 4 |
~1 Citing
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
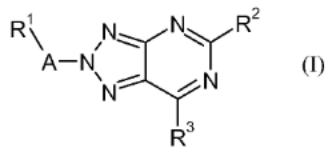
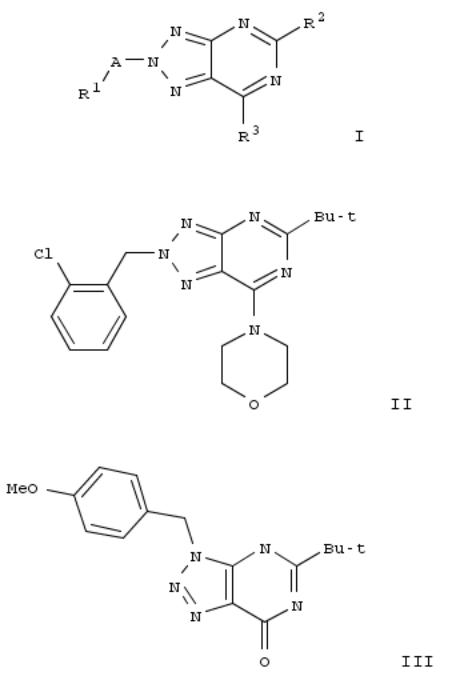
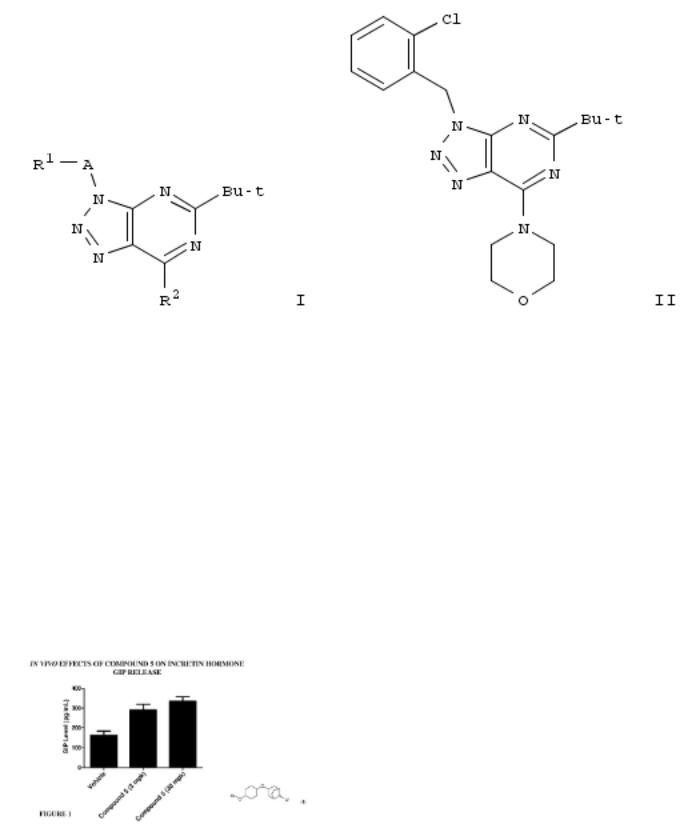
7. Preparation of 1,2,3-triazolo[4,5-d]pyrimidine derivatives as cannabinoid receptor 2 agonists
By Bissantz, Caterina; Grether, Uwe; Kimbara, Atsushi; Nettekoven, Matthias; Roever, Stephan; Rogers-Evans, Mark From PCT Int. Appl. (2013), WO 2013076182 A1 20130530, Language: English, Database: CAPLUS
SciFinder® |
Page 5 |
The invention is related to the prepn. of triazolopyrimidines I [A = alkylene, hydroxyalkylene, CH2CO, CO, SO2, absent; R1 = H, OH, heterocyclyl, haloheteroaryl, etc.; R2 = alkyl, cycloalkoxyalkyl, alkylamino, etc.; R3 = halo, NH2 and derivs.], their pharmaceutically acceptable salts and esters as cannabinoid receptor 2 agonists and to pharmaceutical compns. contg. them. Thus, II was prepd. by a multi-step synthesis starting by azidation of 1-(chloromethyl)-4- methoxybenzene with NaN3, [2+3] cycloaddn. of the azide with 2-cyanoacetamide, acylation of 5-amino-1-(4-methoxybenzyl)- 1H-1,2,3-triazole-4-carboxamide with pivaloyl chloride, intramol. cyclization of the resulting mixt. of 1-(4- methoxybenzyl)-5-pivalamido-1H-1,2,3-triazole-4-carboxamide and N-[4-cyano-1-(4-methoxybenzyl)-1H-1,2,3-triazol-5- yl]pivalamide, chlorination of III with POCl3, amination of 5-tert- butyl-7-chloro-3-(4-methoxybenzyl)-3H-[1,2,3]triazolo[4,5- d]pyrimidine with morpholine, cleavage of the p- methoxyphenylmethyl group and treatment of 5-tert-butyl-7- (morpholin-4-yl)-3H-[1,2,3]triazolo[4,5-d]pyrimidine with 1- (bromomethyl)-2-chlorobenzene. I were tested for their CB2 agonistic activity and selectivity vs. CB1 (data given).
SciFinder® |
Page 6 |
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
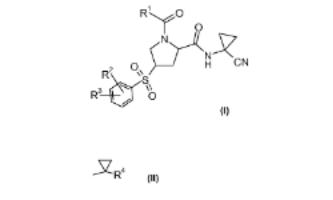
8. Process for the preparation of 1-acyl-4-phenylsulfonylprolinamide derivatives and new intermediates
By Bartels, Bjoern; Bliss, Fritz; Cueni, Philipp; Pfleger, Christophe; Zutter, Ulrich
From PCT Int. Appl. (2013), WO 2013068434 A1 20130516, Language: English, Database: CAPLUS
SciFinder® |
Page 7 |
A novel process for the prepn. of proline derivs. of formula (I; R1 = C1-7 alkyl or Q; R4 = C1-7 alkyl, halogen-C1-7-alkyl, or from Ph optionally substituted by halogen; R2 = halogen or halogen- C1-7-alkyl; R3 = hydrogen, halogen, halogen-C1-7-alkyl, C1-7- alkoxy, halogen-C1-7-alkoxy or a 5- or 6-membered heterocyclic ring contg. one or two nitrogen atoms, the ring which is optionally substituted by C1-7-alkyl or halogen) is described. The proline derivs. I are prepd. by transforming an proline alc. (II; R = OH; R1 = same as above) into the sulfonate II (R = R5SO2O; R5 = C1-7 alkyl, halogen-C1-7-alkyl, or Ph optionally substituted by C1-7 alkyl, nitro, or bromo), reacting the sulfonate with benzene thiols (III; R2, R3 = same as above; R6 = hydrogen or protecting group) to form the thioethers II (R = Q1; R1 , R2, R3 = same as above), and oxidizing the thioethers to form the proline derivs. I. The proline derivs. I are preferential inhibitors of the cysteine protease Cathepsin S and are therefore useful to treat metabolic diseases like diabetes, atherosclerosis, abdominal aortic aneurysm, peripheral arterial disease and diabetic nephropathy (no data). Thus, esterification of (2S,4S)-4-Hydroxy-1-(1- methylcyclopropanecarbonyl)pyrrolidine-2-carboxylic acid (1- cyanocyclopropyl)amide (IV; Ra = H) by benzenesulfonyl chloride in the presence of 4-(dimethylamino)pyridine and Et3N in CH2Cl2 at room temp. for 16 h to give 85% II (Ra = benzenesulfonyl). Thioetherification of the benzenesulfonate II (Ra = benzenesulfonyl) with 4-(2,2,2-trifluoroethoxy)-2- trifluoromethylbenzenethiol in the presence of potassium tertbutoxide in THF at room temp. for 15 h gave 96.1% the thioether (V) which was oxidized by magnesium monoperoxyphthalate hexahydrate in methanol at 50° for 8 h to give 93.3% (2S,4R)-1-(1-methylcyclopropanecarbonyl)-4-[4- (2,2,2-trifluoroethoxy)-2- trifluoromethylbenzenesulfonyl]pyrrolidine-2-carboxylic acid (1- cyanocyclopropyl)amide (VI).
SciFinder® |
Page 8 |
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
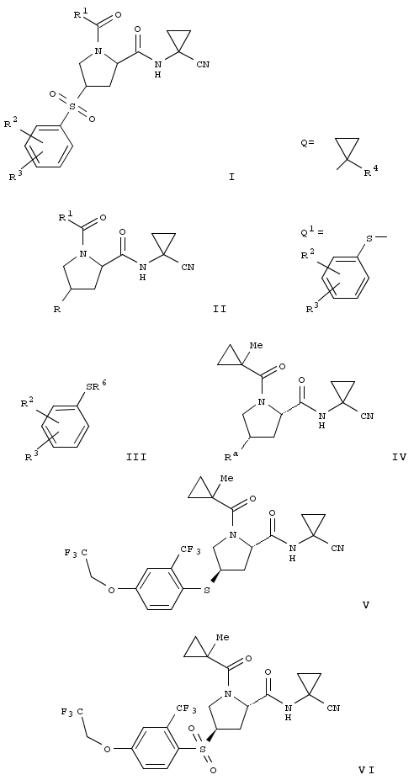
9. Preparation of arylquinoline derivatives as dual FABP inhibitors
By Ceccarelli, Simona M.; Conte, Aurelia; Kuehne, Holger; Kuhn, Bernd; Neidhart, Werner; Obst Sander, Ulrike; Rudolph, Markus
From PCT Int. Appl. (2013), WO 2013064465 A1 20130510, Language: English, Database: CAPLUS
SciFinder® |
Page 9 |
The invention relates to arylquinoline compds. of formula I, as dual FABP 4/5 inhibitors; their prepn. and use in the treatment of various diseases. Compds. of formula I wherein R1 is (un)substituted (halo)alkyl, cycloalkyl, alkoxy, etc.; R2 is H, halo, alkyl, etc.; R3 is H, halo, alkoxy, etc.; R4 is H, NO2, CN, etc.; R5 is H, halo, (un)substituted aminoalkyl, etc.; R6 is (un)substituted Ph and (un)substituted pyridinyl; A is (CH2)0-1; and their pharmaceutically acceptable salts and esters, are claimed. Example II was prepd. by cyclocondensation of (2- amino-5-chlorophenyl)phenylmethanone with Me 3-cyclopropyl- 3-oxopropanoate followed by hydrolysis. All the invention compds. were evaluated for their FABP inhibitory activity.
From the assay, it was detd. that example compd. II exhibited IC50 value of 0.0424 μM and 4.786 μM towards FABP4 and FABP5, resp.
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
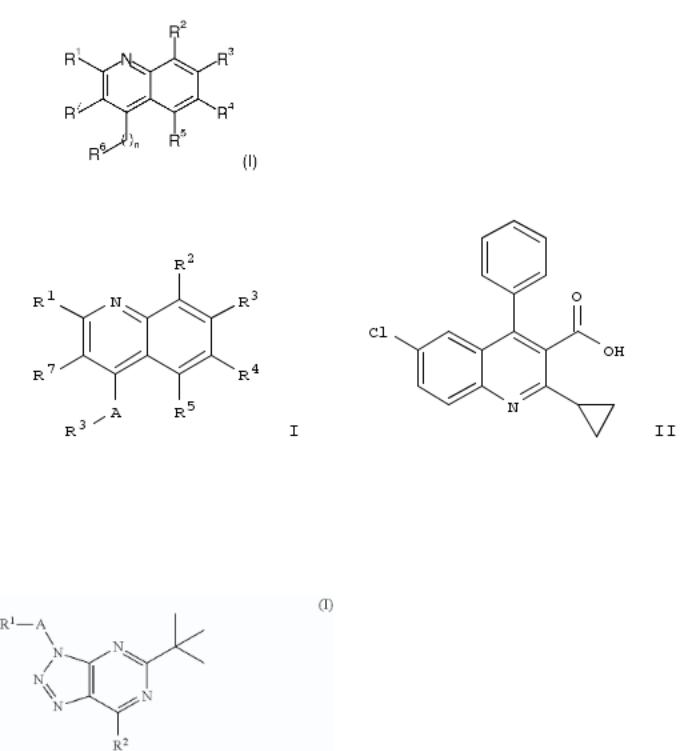
10. Preparation of [1,2,3]triazolo[4,5-d]pyrimidine derivatives useful as cannabinoid receptor 2 agonists
By Adam, Jean-Michel; Bissantz, Caterina; Grether, Uwe; Kimbara, Atsushi; Nettekoven, Matthias; Roever, Stephan; Rogers-Evans, Mark
From U.S. Pat. Appl. Publ. (2013), US 20130116236 A1 20130509, Language: English, Database: CAPLUS
The invention relates to compds. of formula I and their prepn., useful in pharmaceutical compns. esp. as cannabinoid receptor 2 (CB2) agonists. Compds. of formula I, in which R1 is H, (halo)alkyl, OH, etc.; R2 is halo, NH2, OH, and derivs.; A is (hydroxy)alkylene, CH2CO, CO, SO2, and bond; or pharmaceutically acceptable salts and esters thereof, are claimed. Example compd. II was prepd. by cyclocondensation of 5-amino-1-(2-chlorobenzyl)-1H-1,2,3-triazole-4-carboxamide with pivaloyl chloride to yield 5-tert-butyl-3-(2-chlorobenzyl)-3H- [1,2,3]triazolo[4,5-d]pyrimidin-7(4H)-one, which underwent addn. with morpholine to yield II. Invention compds. were tested for their CB2 agonistic activity and selectivity vs. CB1 (data given).
SciFinder® |
Page 10 |
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
11. An efficient procedure for the synthesis of 2,3-unsaturated-O-glycosides: TiCl3(OTf) as the catalyst for type I Ferrier rearrangement
By Chen, Peiran; Lin, Lei
From Tetrahedron (2013), 69(23), 4524-4531. Language: English, Database: CAPLUS, DOI:10.1016/j.tet.2013.04.051
An efficient type I Ferrier rearrangement reaction system for the synthesis of 2,3-unsatd.-O-glycosides has been established by using TiCl3(OTf) as the catalyst. A series of 2,3-unsatd.-O-glucosides were prepd. from 3,4,6-tri-O-acetyl- D-glucal in good yield and high anomeric selectivity.
~1 Citing
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
12. Preparation of cyclohexyloxyazabicyclooctane derivatives for use as GPR119 receptor modulators
By Jones, Robert M.; Buzard, Daniel J.; Han, Sangdon; Kim, Sun Hee; Lehmann, Juerg; Zhu, Xiuwen From PCT Int. Appl. (2013), WO 2013055910 A1 20130418, Language: English, Database: CAPLUS
Title compds. I [A = (un)substituted aryl or heteroaryl; R1 = (un)substituted alkoxycarbonyl, cycloalkylcarbonyl, or heteroaryl], and their pharmaceutically acceptable salts, are prepd. and disclosed as GPR119 receptor modulators. Thus, e.g., II was prepd. by a multistep procedure (prepn. given). Select I were evaluated in hGPR119 activity assays, e.g., II demonstrated an EC50 value of 2.7 nM.