

Reference_01_08_2014_165529
.pdfSciFinder® |
Page 141 |
197. Preparation of heterocyclic substituted phenyl methanones as inhibitors of the glycine transporter 1
By Jolidon, Synese; Narquizian, Robert; Norcross, Roger, David; Pinard, Emmanuel
From PCT Int. Appl. (2006), WO 2006082001 A1 20060810, Language: English, Database: CAPLUS
The title compds. I [R1 = halo, cycloalkyl, aryl, etc.; R2 = SO2(alkyl), SO2NH(alkyl), NO2 or CN; het = arom. or partially arom. bicyclic amine, which may contain one or two addnl. N-atoms, selected from (un)substituted isoindolyl, indolyl, pyrrolo[3,4-d]pyrimidinyl, etc.], useful for treating illnesses based on the glycine uptake inhibitor, were prepd. and formulated. Thus, reacting 2-isopropoxy-5-methansulfonylbenzoic acid (prepn. given) with 2,3-dihydro-1H-isoindole afforded 88% II. It has been found that the compds. I are good inhibitors of the glycine transporter 1 (GlyT-1) (IC50 values were given for representative compds. I).
~4 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
198. Preparation of pyridine derivatives as inhibitors of c-jun n-terminal kinases for the treatment of diabetes and other diseases
By Liu, Gang; Sham, Hing L.; Szczepankiewicz, Bruce G.; Xin, Zhili; Zhao, Hongyu; Serby, Michael D.; Liu, Bo; Liu, Mei
From U.S. Pat. Appl. Publ. (2006), US 20060173050 A1 20060803, Language: English, Database: CAPLUS
Title compds. I [wherein L = -C(O)-, -NH-, -C(NH)-NH-, etc.; R1 = alkenyl, alkenyloxyalkyl, alkoxyalkoxyalkyl, etc.; R2, R4 = H, alkyl, alkylcarbonyl, etc.; R3 = H, alkyl, azido, halo, etc.; R5 = alkenyl, alkoxy, alkyl, etc.] and pharmaceutically acceptable prodrugs and salts thereof were prepd. as inhibitors of c-jun n-terminal kinases (JNK). For instance, substitution of 2-bromo-4,6-diaminonicotinonitrile with EtONa under microwave heating (65% yield) followed by N- acylation with acetyl chloride (50% yield) gave N-pyridinylacetamide II. I were found to inhibit the activities of JNK1 and JNK2 with IC50 in a range of about 0.001 μM to about 10 μM. Therefore, I and their pharmaceutical compns. are useful for the prevention or treatment of disorders regulated by the activation of JNK1, JNK2 and JNK3, such as diabetes.
~4 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
199. Preparation of benzoylpiperazine derivatives as glycine transporter 1 (GlyT-1) inhibitors

SciFinder® |
Page 142 |
By Jolidon, Synese; Narquizian, Robert; Norcross, Roger David; Pinard, Emmanuel
From PCT Int. Appl. (2006), WO 2006072436 A1 20060713, Language: English, Database: CAPLUS
Title compds. represented by the formula I [wherein R1 = OR, SR or heterocycloalkyl; R = (halo)alkyl or -(CH2)n- cycloalkyl; n = 0-2; R2 = -SO2-alkyl, -SO2NH-alkyl, NO2 or CN; X1, X2 = independently CR' or N; R' = independently H, halo, alkyl, CN, etc.; and pharmaceutically acceptable acid addn. salts thereof] were prepd. as glycine transporter 1 (GlyT-1) inhibitors. For example, reaction of 5-methylsulfonyl-2-(2,2,3,3,3-pentafluoropropoxy)benzoic acid (prepn. given) with 1-(5-trifluoromethylthiazol-2-yl)piperazine•HCl gave II in 68% yield. Thus, II showed glycine uptake inhibition with an IC50 value of 0.006 μM. Thus, I are useful as GlyT-1 inhibitors for the treatment of neurol. and neuropsychiatric disorders, such as schizophrenia (no data).
~3 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
200. meso-Trifluoromethyl-substituted expanded porphyrins
By Shimizu, Soji; Aratani, Naoki; Osuka, Atsuhiro
From Chemistry - A European Journal (2006), 12(18), 4909-4918. Language: English, Database: CAPLUS, DOI:10.1002/chem.200600158
A series of meso-trifluoromethyl-substituted expanded porphyrins, including N-fused [24]pentaphyrin, [28]hexaphyrin, [32]heptaphyrin, [46]decaphyrin, and [56]dodecaphyrin, were synthesized by means of an acid-catalyzed one-pot condensation reaction of 2-(2,2,2-trifluoro-1-hydroxyethyl)pyrrole as the first examples bearing meso-alkyl substituents. Besides these products, meso-tetrakis(trifluoromethyl)porphyrin and two calix[5]phyrins were also obtained. [28]Hexaphyrin was quant. oxidized to [26]hexaphyrin with MnO2. These expanded porphyrins have been characterized by mass spectrometry, 1H and 19F NMR spectroscopy, and UV/Vis spectroscopy. The single-crystal structures have been detd. for five of the compds. The N-fused [24]pentaphyrin displays a distorted structure contg. a tricyclic fused moiety that is similar to those of meso-aryl-substituted counterparts, whereas the calix[5]phyrins are indicated to take roughly planar conformations with an inverted pyrrole opposite to the sp3-hybridized meso-carbon atom. Both [28]- and [26]hexaphyrins have figure-eight structures. Solid-state structures of the decaphyrin and dodecaphyrin are remarkable, exhibiting a crescent conformation and an intramol. two-pitch helical conformation, resp.
~48 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
201. Discovery of 3-arylpropionic acids as potent agonists of sphingosine-1-phosphate receptor-1 (S1P1) with high selectivity against all other known S1P receptor subtypes
SciFinder® |
Page 143 |
By Yan, Lin; Huo, Pei; Doherty, George; Toth, Lesile; Hale, Jeffrey J.; Mills, Sander G.; Hajdu, Richard; Keohane, Carol A.; Rosenbach, Mark J.; Milligan, James A.; et al
From Bioorganic & Medicinal Chemistry Letters (2006), 16(14), 3679-3683. Language: English, Database: CAPLUS, DOI:10.1016/j.bmcl.2006.04.084
A series of 3-arylpropionic acids were synthesized as S1P1 receptor agonists. Structure-activity relationship studies on the pendant Ph ring revealed several structural features offering selectivity of S1P1 binding against S1P2-5. These highly selective S1P1 agonists induced peripheral blood lymphocyte lowering in mice and one of them was found to be efficacious in a rat skin transplantation model, supporting that S1P1 agonism is primarily responsible for the immunosuppressive efficacy obsd. in preclin. animal models.
~16 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
202. Reactions of aliphatic fluoro-alcohols with CHClF2 at atmospheric pressure
By Mizukado, Junji; Matsukawa, Yasuhisa; Quan, Heng-dao; Tamura, Masanori; Sekiya, Akira
From Journal of Fluorine Chemistry (2006), 127(3), 400-404. Language: English, Database: CAPLUS, DOI:10.1016/j.jfluchem.2005.12.034
The reactions of aliph. fluoro-alcs. with chlorodifluoromethane (CHClF2) at atm. pressure were examd. In the reaction of CF3CF2CH2OH, the difluoromethylated ether was obtained in moderate yield by using ethers such as 1,4-dioxane, diglyme, and THF, or their mixts. with water as a reaction solvent. While acetal and orthoformate were also produced, the selectivity of the difluoromethylated ether could be improved by adding water to the reaction. The effect of water could be explained by the reaction mechanism.
~1 Citing
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
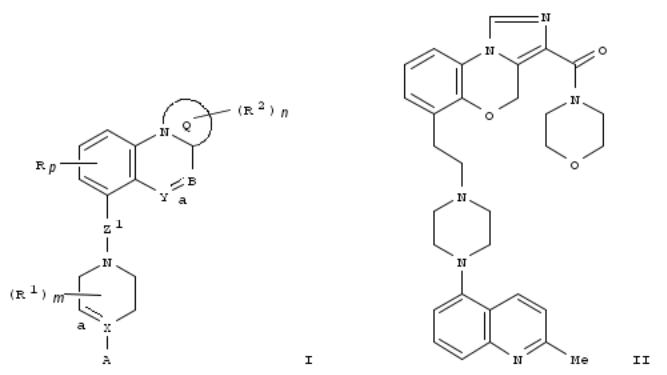
203. Preparation of fused tricyclic imidazobenzoxazines, imidazoquinolines, triazolobenzoxazines and their analogs for the treatment of psychotic disorders and related diseases
By Bentley, Jonathan; Bergauer, Markus; Bertani, Barbara; Biagetti, Matteo; Borriello, Manuela; Bromidge, Steven Mark; Gianotti, Massimo; Granci, Enrica; Leslie, Colin Philip; Pasquarello, Alessandra; et al
From PCT Int. Appl. (2006), WO 2006024517 A1 20060309, Language: English, Database: CAPLUS
Fused tricyclic compds. I [wherein a = single or double bond; ring Q = (un)substituted 5-membered heteroaryl or heterocyclyl; B = (un)substituted CH or CH2; Y = (un)substituted CH2, O, etc.; Z1 = ethylene, etc.; X = CR1 or N when a is a single bond; X = C when a is a double bond; A = (un)substituted indolyl, quinolyl, benzofuranyl, etc.; R = halo, alkyl, cyano, etc.; R1 = H, halo, alkyl, etc.; R2 = H, halo, hydroxy, etc. p = 0-2; m, n = 0-3] and salts or prodrugs thereof, which possess high affinity for 5-HT1 type receptors and/or are serotonin reuptake inhibitors, were prepd. For instance, imidazobenzoxazine carboxamide II was synthesized in 33% yield by condensation of the corresponding acid (prepn. given) with morpholine in DMF in the presence of TBTU and DIPEA. In a functional potency assay, II had fpKi of 9.7 against 5-HT1A. Therefore, the invented compds. are useful for treating or preventing diseases or conditions mediated by modulation of 5-HT1 receptors and/or serotonin reuptake receptors, such as psychotic disorders.
SciFinder® |
Page 144 |
~12 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
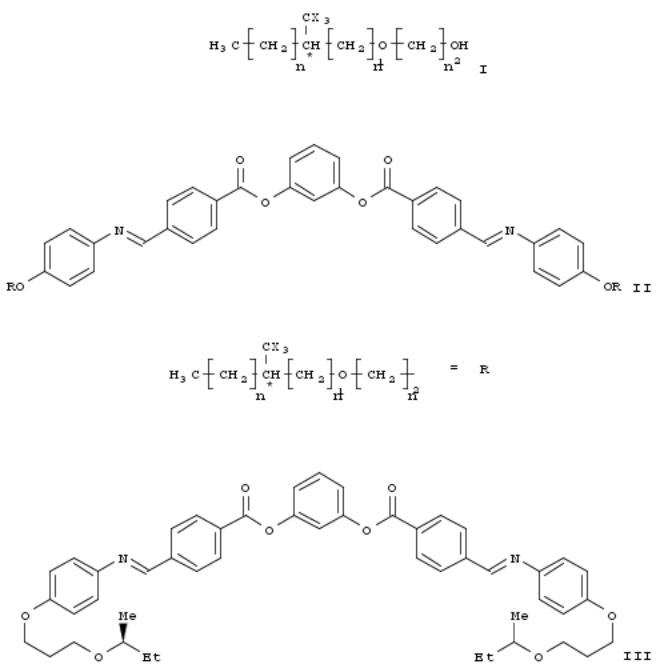
204. Chiral alkoxy alcohols and chiral 1,3-phenylene-bis[4-(4-alkoxy)phenyliminomethyl]benzoates and processes for preparation thereof and their phase transition temperatures
By Kang, Kyung-Tae; Lee, Seng-Kue; Heo, Sung; Park, Chang-Won; Choi, Jin-Wook; Baek, Myeong-Ki; Lee, Sun-Hee From PCT Int. Appl. (2006), WO 2006025687 A1 20060309, Language: English, Database: CAPLUS
The invention relates to chiral alkoxy alcs. of formula I, chiral 1,3-phenylene bis[4-(4- alkoxy)phenyliminomethyl]benzoates of formula II, and the prepn. methods of them. Compds. of formula I and II wherein X is H or F; n is an integer 1 to 5; n1 is an integer 3 to 10; n2 is an integer 0 or 1; * depicts an asym. carbon; and their methods for prepn. are claimed in this invention. In particular, the chiral alkoxy alc. of the invention can be used for synthesizing chiral liq. crystal materials, pharmaceuticals, agricultural chems., and as intermediates for natural product synthesis. Example compd. compd. III was prepd. by mesylation of (S)-3-(but-2-yloxy)propan-1-ol with mesyl chloride and the resulting mesylate was brominated to give the alkoxypropyl bromide, which underwent etherification with 4- hydroxyacetanilide to give (S)-4-[3-(but-2-yloxy)propoxy]acetanilide, which was deacetylated to give the aniline deriv., which underwent condensation with 1,3-phenylene bis(4-formylbenzoate) to give compd. III. The invention compds. of formula II were evaluated for their phase transition temps. Example compd. III had phase transition temps. for going from isotropic liq. phase to liq. crystal phase at 115.4 °C and from liq. crystal phase to crystal phase at 104.4 °C. All the other invention compds. of formula II showed similar phase transition temps. Since the chiral compds. of 1,3-phenylene bis[4-(4-alkoxy)phenyliminomethyl]benzoate have a lower phase transition temp. and exhibit ferroelectricity or antiferroelectricity depending on the length of the chiral (alkoxy)alkyl terminal group, they can be used as an important component in smectic liq. crystal mixts.
SciFinder® |
Page 145 |
~0 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
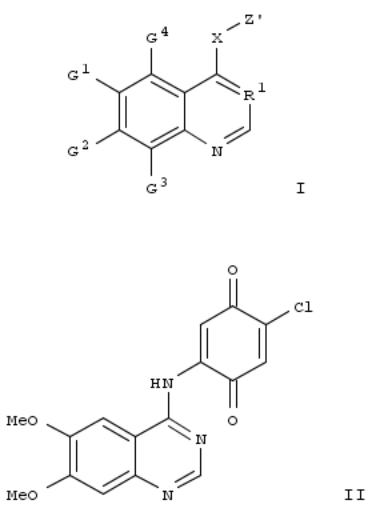
205. Preparation of quinone substituted quinazoline and quinoline kinase inhibitors for treatment of angiogenesis-related diseases
By Floyd, Middleton B., Jr.; Nittoli, Thomas; Wissner, Allan; Dushin, Russell George; Nilakantan, Ramaswamy; Ingalls, Charles; Fraser, Heidi Leigh; Johnson, Bernard Dean
From PCT Int. Appl. (2005), WO 2005115145 A2 20051208, Language: English, Database: CAPLUS
SciFinder® |
Page 146 |
Title compds. I [R1 = N, C-CN, CH, C-F, C-Cl, C,Br, C-I; G1-G4 = independently H, halo, alk(en/yn)yl, alkylsulfinyl, NH2 and derivs., etc., with the proviso that G3 or G4 are not -NH-R2; R2 = -CO-C≡C-R3, -CO-(R3)C:C(R3)2, etc.; R3 = independently H, alkyl, Ph, carboxy, etc.; X = NH, O, S, etc.; Z' = (un)substituted 1,4-benzoquinone, 1,4-naphthoquinone, 7-oxabicyclo[4.1.0]hept-3-ene-2,5-dione; and their pharmaceutically acceptable salts] were prepd. as protein kinases, particularly protein tyrosine kinases, inhibitors. I are useful for treatment of diseases that are characterized, at least in part, by excessive, abnormal, or inappropriate angiogenesis, such as cancer, diabetic retinopathy, macular degeneration and rheumatoid arthritis. I inhibit angiogenesis by inhibiting a tyrosine kinase receptor enzyme, specifically KDR, and binding to the KDR in an irreversible manner. For example, reacting 2-amino-4,5-dimethoxybenzonitrile with DMF di-Me acetal, refluxing of amidine with 4-chloro-2,5-dimethoxyaniline and oxidn. of dimethoxy intermediate with ceric
ammonium nitrate gave quinazoline II. Quinazoline II (100 nM concn.) gave 83% inhibition of KDR kinase activity. Selected I were effective inhibitors of VEGF-dependent growth factor of HUVEC cells.
~1 Citing
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
206. 2-(Quinazolin-4-ylamino)-[1,4]benzoquinones as Covalent-Binding, Irreversible Inhibitors of the Kinase Domain of Vascular Endothelial Growth Factor Receptor-2
By Wissner, Allan; Floyd, M. Brawner; Johnson, Bernard D.; Fraser, Heidi; Ingalls, Charles; Nittoli, Thomas; Dushin, Russell G.; Discafani, Carolyn; Nilakantan, Ramaswamy; Marini, Joseph; et al
From Journal of Medicinal Chemistry (2005), 48(24), 7560-7581. Language: English, Database: CAPLUS, DOI:10.1021/jm050559f

SciFinder® |
Page 147 |
A series of 2-(quinazolin-4-ylamino)-[1,4]benzoquinone derivs. that function as potent covalent-binding, irreversible inhibitors of the kinase domain of vascular endothelial growth factor receptor-2 (VEGFR-2) has been prepd. by ceric ammonium nitrate oxidn. of substituted (2,5-dimethoxyphenyl)(6,7- disubstituted-quinazolin-4-yl)amines and by displacement of the chlorine atom of substituted 2-chloro-5-(6,7-disubstituted- quinazolin-4-ylamino)-[1,4]benzoquinones with various amines, anilines, phenols, and alcs. Enzyme studies were conducted in the absence and presence of glutathione and plasma. Several of the compds. inhibit VEGF-stimulated autophosphorylation in intact cells. Kinetic expts. were performed to study the reactivity of selected inhibitors toward glutathione. Reactivities correlated with LUMO energies calcd. as avs. of those of individual conformers weighted by the Boltzmann distribution. These results and mol. modeling were used to rationalize the biol. observations. The compds. behave as non-ATP- competitive inhibitors. Unequivocal evidence, from mass spectral studies, indicates that these inhibitors form a covalent interaction with Cys-1045. One member of this series displays antitumor activity in an in vivo model.
~38 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
207. Preparation of substituted pyridine alkynes with MCH antagonistic activity for the treatment of metabolic disorders
By Stenkamp, Dirk; Mueller, Stephan Georg; Lustenberger, Philipp; Lehmann-Lintz, Thorsten; Roth, Gerald Juergen; Rudolf, Klaus; Schindler, Marcus; Thomas, Leo; Lotz, Ralf
From U.S. Pat. Appl. Publ. (2005), US 20050234101 A1 20051020, Language: English, Database: CAPLUS
Various substituted pyridinyl alkynes are prepd. For instance, 2-[[4-[[5-(4-chlorophenyl)pyridin-2-yl]ethynyl]-2- methylphenyl]oxy]ethyl methanesulfonate (I) is prepd. in 6 steps from 4-iodophenol, 2-bromoethanol, trimethylsilylacetylene, 2,5-dibromopyridine and 4-chlorophenylboronic acid. This intermediate is reacted with a variety of amines to produce example compds. I is converted to II by displacement with the corresponding amine. II exhibits an IC50 = 6.2 nM for MCH-1. Example compds. are useful for the treatment of metabolic disorders and/or eating disorders, particularly obesity and diabetes.
~1 Citing
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
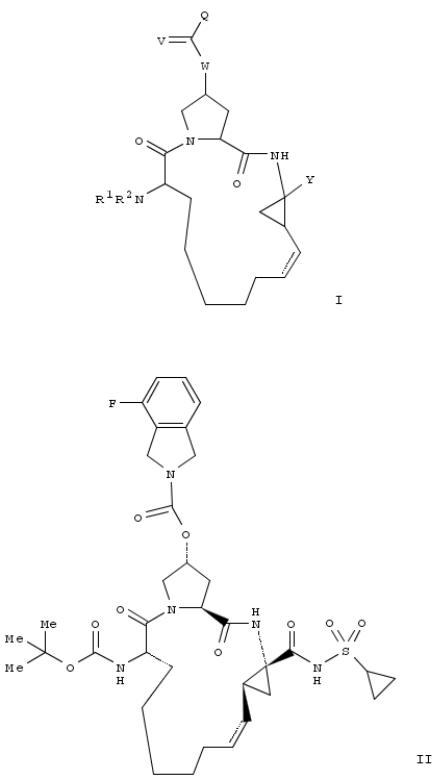
208. Preparation of macrocyclic carboxylic acids or sulfonamides as inhibitors of HCV replication
SciFinder® |
Page 148 |
By Blatt, Lawrence M.; Wenglowsky, Steven M.; Andrews, Steven W.; Condroski, Kevin R.; Jiang, Yutong; Kennedy, April L.; Doherty, George A.; Josey, John A.; Stengel, Peter J.; Woodard, Benjamin T.; et al
From PCT Int. Appl. (2005), WO 2005095403 A2 20051013, Language: English, Database: CAPLUS
The invention relates to macrocyclic compds., e.g., I [Q is (un)substituted 2-isoindolinyl, 2-isoquinolinyl, 1- benzoazetidinyl, 1-indolinyl, (3,4-dehydro)pyrrolidino, (3,4-dehydro)piperidino or Q is R3-R4, where R3 is alkyl, cycloalkyl, alkylcycloalkyl, Ph, pyridyl and other heterocyclic groups and R4 is H, Ph, pyridyl and other heterocyclic groups; V is O, S, NH; W is O, NR5 or CR5, where R5 is H, alkyl, fluoroalkyl, cycloalkyl, alkylcycloalkyl; Y is a sulfonimide CONHSO2R6, where R6 is (un)substituted alkyl, fluoroalkyl, cycloalkyl, alkylcycloalkyl, aryl, heteroaryl or (un)substituted phenyl; or Y is carboxy or a pharmaceutically-acceptable salt or prodrug; R1 is H, (un)substituted alkyl, cycloalkyl, alkylcycloalkyl, Ph or benzyl; R2 is H, alkyl, (thio)carbamoyl, acyl, or sulfonyl group; the dashed line represents an optional double bond], for use in pharmaceutical compns. for the treatment of flaviviral or hepatitis C virus (HCV) infection and liver fibrosis. Thus, compd. II, prepd. by reaction of the macrocyclic prolinol deriv. with CDI and 4-fluoro-2,3-dihydro-1H-isoindole, showed IC50 and EC50 < 0.1 μM in the NS3-NS4A protease inhibition assay and did not display toxicity in Rattus sp. when dosed for seven days at 30 mg/kg BID, providing at least a 10-fold safety margin above the presumptive efficacious dose (3 mg/kg) that yields liver concns. 100-fold in excess of the replicon EC50 value of the compd.
SciFinder® |
Page 149 |
~15 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
209. Unexpected and expeditious entry into trifluoromethyl aziridines from substituted β-dicarbonyl compounds
By Colantoni, Daniele; Fioravanti, Stefania; Pellacani, Lucio; Tardella, Paolo A.
From Journal of Organic Chemistry (2005), 70(23), 9648-9650. Language: English, Database: CAPLUS, DOI:10.1021/jo0515327
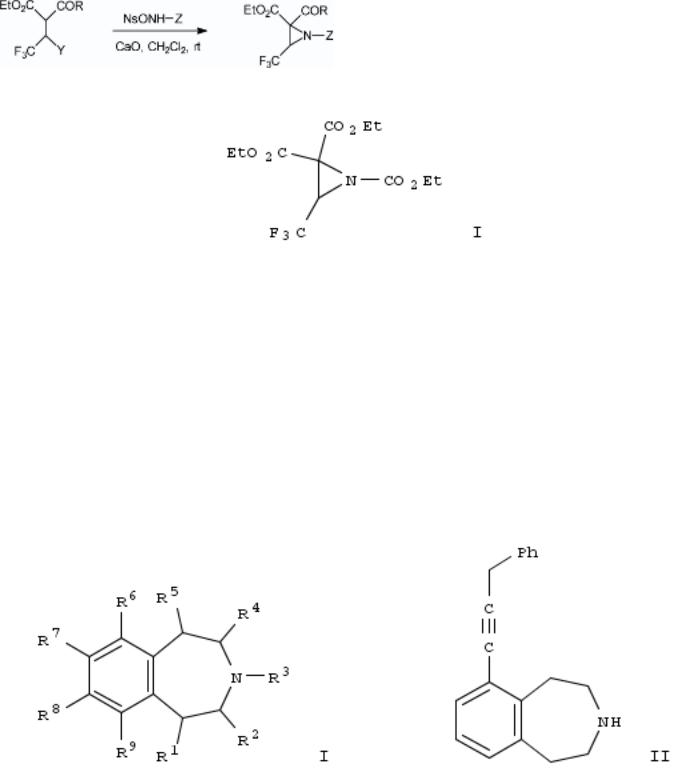
SciFinder® |
Page 150 |
One-pot aziridinations were obtained starting from substituted 2,2,2-trifluoroethyl β-dicarbonyl compds. with nosyloxycarbamates in the presence of an excess of CaO as base. The unexpected ring closure reaction took place at room temp., leading to the N-protected α-trifluoromethyl aziridines, e.g., I, with good yields. The reaction pathway seems to be influenced by the choice of the base.
~11 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
210. Preparation of 6-substituted 2,3,4,5-tetrahydro-1H-benzo[d]azepines as 5-HT2c receptor agonists
By Allen, John Gordon; Briner, Karin; Cohen, Michael Philip; Galka, Christopher Stanley; Hellman, Sarah Lynne; Martinez-Grau, Maria Angeles; Reinhard, Matthew Robert; Rodriguez, Michael John; Rothhaar, Roger Ryan; Tidwell, Michael Wade; et al
From PCT Int. Appl. (2005), WO 2005082859 A1 20050909, Language: English, Database: CAPLUS
The title compds. I [R1 = H, F, alkyl; R2-R4 = H, Me, Et; R5 = H, F, Me, Et; R6 = C≡CR10, OR12, SR14, substituted NH2; R7 = H, halo, CN, etc.; R8 = H, halo, CN, SCF3; R9 = H, halo, CN, etc.; R10 = CF3, alkyl, fluoroalkyl, etc.; R12 = pyridylalkyl, thiazolylalkyl, etc.; R13 = cycloalkylalkyl, alkoxy, fluoroalkoxy, etc.; R14 = tetrahydropyranyl, tetrahydrofuranyl, etc.], useful as selective 5-HT2c receptor agonists for the treatment of 5-HT2c assocd. disorders including obesity, obsessive/compulsive disorder, depression, and anxiety, were prepd. Thus, reacting 3-tert-butoxycarbonyl-6- trifluoromethanesulfonyloxy-2,3,4,5-tetrahydro-1H-benzo[d]azepine (prepn. given) with 3-phenyl-1-propyne followed by deprotection afforded 85% II.HCl. Representative compds. I are found to have excellent affinity for the 5-HT2c receptor, with Ki's typically less than or equal to about 200 nM.
~6 Citings
Copyright © 2014 American Chemical Society (ACS). All Rights Reserved.
211. Towards the syntheses of α-trifluoromethylated oxygenated heterocycles and their precursors
By Harthong, Steven; Billard, Thierry; Langlois, Bernard R.
From Synthesis (2005), (13), 2253-2263. Language: English, Database: CAPLUS, DOI:10.1055/s-2005-872076