

7 семестр / Основы_физич_химии_Теория_и_задачи_Еремин_и_др_2005_480с
.pdf
Г л а в а 5. Химическая кинетика |
365 |
В рамках этой теории число внутренних степеней свободы f является эмпирическим параметром и определяется по экспериментальным данным.
Схема Линдемана послужила основой для многих более сложных теорий мономолекулярных реакций. Все теории этого типа основаны на учете зависимости констант скорости активации и распада от энергии, при этом рассматривается активация в малый интервал энергий от E до
E + δE:
A + M → A(E→ E + δE ) + M , |
(25.19.а) |
|
δk1 |
* |
|
A(E→ E + δE) + M → A + M , |
(25.19.б) |
|
* |
k2 |
|
A(E ) |
→ P , |
(25.19.в) |
* |
ka (E) |
|
Применение квазистационарного приближения к этой схеме дает выражение, аналогичное (25.16), для вклада активных молекул с энергией от E до E + δE в константу скорости:
|
ka (E) |
δk1(E→ E + δE) |
|
|
|||
δkI(E→ E + δE ) = |
|
k2 |
. |
(25.20) |
|||
|
|
||||||
1 + |
ka (E) |
||||||
|
|
|
|||||
|
k2 [M] |
|
|
|
|||
Полная константа скорости мономолекулярной реакции получается интегрированием (25.20) по всем возможным энергиям активации:
|
ka (E) |
dk1(E→ E + δE) |
|
|
∞ |
k2 |
|
|
|
kI = ∫ |
|
. |
(25.21) |
|
|
ka (E) |
EA 1 + k2 [M]
В теории РРКМ (Райса–Рамспергера–Касселя–Маркуса), вводится различие между активной молекулой A* и активированным комплексом A+, и реакция (25.19.в) записывается в две стадии:
A |
* |
ka (E ) |
+ |
k + |
(25.22) |
(E ) |
→ A |
|
→ P . |
Активная молекула обладает достаточной энергией, чтобы вступить в реакцию, однако распределение энергии в молекуле не позволяет ей сразу превратиться в реагенты, для этого сначала должна быть достигнута конфигурация активированного комплекса. В теории РРКМ константу скорости k1 рассчитывают с помощью квантовых методов, а константу ka – по теории активированного комплекса.
366 |
Г л а в а 5. Химическая кинетика |
|
|
|
|
|
|
|||||
|
Теория активированного комплекса |
|
|
|
||||||||
|
Современные теории химической кинетики основаны на анализе |
|||||||||||
|
движения ядер по поверхности потенциальной энергии системы реаги- |
|||||||||||
|
рующих частиц. Для описания химической реакции решают классиче- |
|||||||||||
|
ские или квантовые уравнения движения, а константы скорости находят |
|||||||||||
|
из полученных решений – классических траекторий, волновых функ- |
|||||||||||
|
ций, функций распределения и т.д. |
|
|
|
|
|
|
|
|
|
|
|
|
Для системы, содержащей N атомов, потенциальная энергия зависит |
|||||||||||
|
|
от n = 3N – 6 (для нелинейной |
||||||||||
|
D |
системы) или n = 3N – 5 (ли- |
||||||||||
|
|
нейная молекула) координат и |
||||||||||
|
|
может |
быть |
представлена ги- |
||||||||
2 |
|
перповерхностью |
размерности |
|||||||||
X-Y |
B |
|||||||||||
n + 1. Для наглядного представ- |
||||||||||||
|
|
|||||||||||
|
|
ления |
этой |
|
гиперповерхности |
|||||||
|
|
используют |
|
сечения |
|
вдоль |
||||||
|
|
координат, |
существенных для |
|||||||||
|
C |
протекающей реакции. Напри- |
||||||||||
A |
|
мер, при диссоциации линей- |
||||||||||
O |
ной |
трехатомной |
молекулы |
|||||||||
|
Расстояние Y1-X |
YXY → YX + Y потенциаль- |
||||||||||
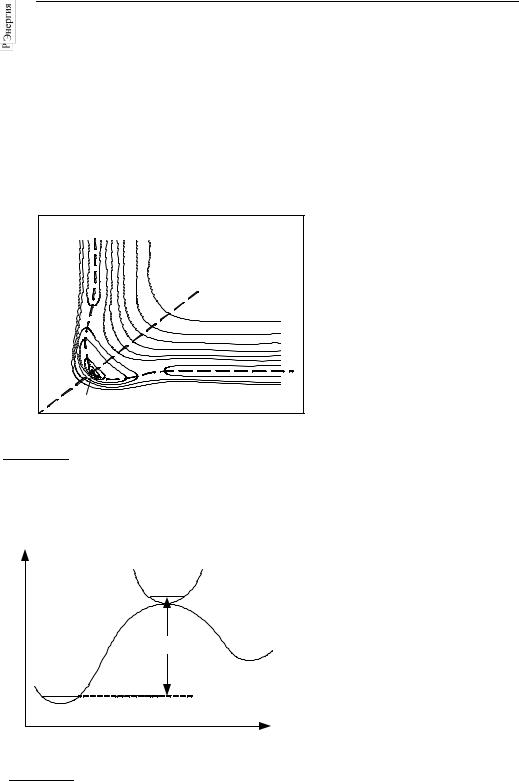
Рис. 25.1 |
Поверхность потенциальной энергии |
ную |
энергию можно |
|
предста- |
|||||||
вить как функцию двух межъя- |
||||||||||||
|
для симметричной трехатомной |
|||||||||||
|
дерных |
|
расстояний, |
для |
||||||||
|
молекулы YXY. Симметричное |
|
||||||||||
|
колебание происходит вдоль линии AB, |
изображения |
которой |
исполь- |
||||||||
|
а асимметричное, приводящее к |
зуют линии уровня (рис. 25.1). |
||||||||||
|
распаду, – вдоль линий OC или OD. |
Одномерную кривую, ха- |
||||||||||
|
|
|||||||||||
|
|
рактеризующую путь с наи- |
||||||||||
|
(Y1···XY2)‡ |
меньшей энергией, по которому |
||||||||||
|
|
происходит реакция, |
называют |
|||||||||
|
|
координатой реакции. |
Сечение |
|||||||||
|
|
поверхности вдоль этой кривой |
||||||||||
|
E0 |
дает |
профиль |
потенциальной |
||||||||
|
энергии для реакции (рис. 25.2), |
|||||||||||
|
C |
на котором имеется максимум. |
||||||||||
|
|
Все конфигурации ядер, соот- |
||||||||||
O |
|
ветствующие |
малой |
|
области |
|||||||
|
вблизи этого максимума, назы- |
|||||||||||
|
|
|||||||||||
|
Координата реакции |
вают |
|
активированным |
ком- |
|||||||
|
плексом, |
или |
|
переходным |
||||||||
Рис. 25.2 |
Профиль потенциальной энергии |
|
||||||||||
состоянием |
|
для |
|
данной |
реак- |
|||||||
|
для реакции YXY → Y + XY. |
ции. Активированный комплекс |
||||||||||
|
Координата реакции – расстояние |
|||||||||||
|
можно |
рассматривать |
как |
|||||||||
|
вдоль линии OC на рис. 25.1 |
|||||||||||
|
обычную молекулу с той раз- |
|||||||||||
|
|
|||||||||||




372 |
Г л а в а 5. Химическая кинетика |
ПРИМЕРЫ
Пример 25-1. Константа скорости бимолекулярной реакции
2NO2 → 2NO + O2
при 627 °С равна 1.81 103 см3 моль–1 с–1. Вычислите истинную энергию активации и долю активных молекул, если радиус молекулы NO2 можно принять равным 3.55 Å, а стерический множитель для этой реакции равен 0.019.
Решение. При расчете будем опираться на теорию активных столк-
новений (формулы (25.5), (25.8)):
|
− |
EA |
|
= |
|
|
|
k(T ) |
|
|
|
|
|
= |
||
exp |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
RT |
|
|
|
|
|
|
|
|
|
1 / 2 |
|
|||||
|
|
|
|
8P N |
|
r |
2 |
|
πRT |
|
|
|||||
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
A |
X |
|
|
M |
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
= |
|
|
1.81 10−3 |
|
|
|
|
= 2.20 |
10−10. |
||
|
23 |
|
−10 |
|
2 |
|
3.14 8.31 900 1 / 2 |
||||
|
|
|
|
|
|
||||||
|
8 0.019 6.02 10 |
|
(3.55 10 |
|
) |
|
|
|
|
|
|
|
|
|
0.046 |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
Это число представляет собой долю активных молекул.
При расчетах констант скорости с использованием различных теорий химической кинетики необходимо очень аккуратно обращаться с размерностями. Проще всего приводить все единицы к системе СИ.
Истинную энергию активации легко вычислить через долю активных молекул:
EA = −RT ln(2.20 10−10 ) =8.31 900 22.2 =166000 Дж моль–1 = 166 кДж моль–1.
Пример 25-2. Используя теорию активированного комплекса, определите температурную зависимость константы скорости тримолекулярной реакции
2NO + Cl2 = 2NOCl
при температурах, близких к комнатной. Найдите связь между опытной и истинной энергиями активации.
Решение. Согласно статистическому вариант ТАК, константа скорости равна (формула (25.24)):
|
kBT |
|
Q |
≠ |
|
|
|
E |
≠ |
|
k(T ) = |
(N AV )2 |
|
|
exp |
− |
0 |
. |
|||
h |
QNO2 QCl |
|
|
|
||||||
|
|
2 |
|
|
RT |
|||||
|
|
|
|
|
|
|
|
|
|
|
В суммах по состояниям активированного комплекса и реагентов мы не будем учитывать колебательные и электронные степени свободы,
Г л а в а 5. Химическая кинетика |
373 |
т.к. при низких температурах колебательные суммы по состояниям близки к единице, а электронные – постоянны.
Температурные зависимости сумм по состояниям с учетом поступательного и вращательного движений имеют вид:
Q≠ = Qпост Qвращ ~ T 3/ 2 T 3/ 2 = T 3 ,
QNO = Qпост Qвращ ~ T 3/ 2 T = T 5 / 2 ,
QCl2 = Qпост Qвращ ~ T 3/ 2 T = T 5 / 2 .
Активированный комплекс (NO)2Cl2 – нелинейная молекула, поэтому его вращательная сумма по состояниям пропорциональна T 3/2.
Подставляя эти зависимости в выражение для константы скорости, находим:
|
|
|
T 3 |
|
|
E0≠ |
|
−7 / 2 |
|
E0≠ |
||
k(T ) ~ T |
|
|
|
|
exp − |
|
|
= T |
|
exp − |
|
. |
|
|
2 |
|
|
|
|
||||||
|
|
5 / 2 |
5 / 2 |
|
RT |
|
|
|
RT |
|||
|
(T |
) T |
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
||
Мы видим, что тримолекулярные реакции характеризуются довольно необычной зависимостью константы скорости от температуры. При определенных условиях константа скорости может даже убывать с ростом температуры за счет предэкспоненциального множителя!
Опытная энергия активации этой реакции равна:
|
2 d ln k(T ) |
≠ |
|
7 |
|
|
Eоп = RT |
|
|
= E0 |
− |
|
RT . |
|
dT |
2 |
||||
|
|
|
|
|
||
Пример 25-3. Используя статистический вариант теории активированного комплекса, получите выражение для константы скорости мономолекулярной реакции.
Решение. Для мономолекулярной реакции A A≠ → продукты
константа скорости, согласно (25.24), имеет вид:
|
kBT |
|
Q ≠ |
|
|
E0≠ |
|
k(T ) = |
|
|
|
exp |
− |
|
. |
h |
QA |
|
|||||
|
|
|
|
RT |
|||
|
|
|
|
|
|
|
|
Активированный комплекс в мономолекулярной реакции представляет собой возбужденную молекулу реагента. Поступательные суммы
реагента A и комплекса A≠ одинаковы (масса – одна и та же). Если предположить, что реакция происходит без электронного возбуждения, то и электронные суммы по состояниям одинаковы. Если считать, что при возбуждении строение молекулы реагента изменяется не очень