

Технологии органического и нефтехимического синтеза
..pdfиз этилена и СО в среде воды в присутствии катализатора СоI2 или Со(АсО)2 с добавкой C2H5I (отношение I : Со = (2,5–3,5) : 1). Предложен способ одновременного получения уксусной и пропионовой кислот карбонилированием этилена в водном метаноле под действием соединений родия и галоидного промотора при температуре 175– 200 °С и давлении до 7 МПа. Эффективными катализаторами синтеза пропионовой и жирных кислот С17–С30 и их эфиров из олефинов, СО и воды или спирта в мягких условиях (температура 80–150 °С, давление 4–15 МПа) являются комплексы Со2(СО)8 с пиридиновыми основаниями, а также комплексы Pd или Pt. Условия получения пропионовой кислоты карбонилирования этилена приведены в табл. 11.2.
Таблица 1 1 . 2
Промышленные и перспективные способы получения пропионовой кислоты карбонилированием этилена
Фирма |
Катализатор |
Температура, |
Давление, |
Выход |
|
|
°С |
МПа |
пропионовой |
|
|
|
|
кислоты, мас. % |
Du Pont |
Карбонил, ацетат, |
280 |
30–35 |
89 |
|
пропионат никеля |
|
|
|
Shell |
Н3РО4 + BF3 |
20–80 |
5–10 |
80 |
BASF |
Карбонил никеля |
230–320 |
30–40 |
95 |
Union Oil |
Соли родия |
170 |
10 |
– |
Monsanto |
Комплексы иридия |
191 |
4,9 |
65 |
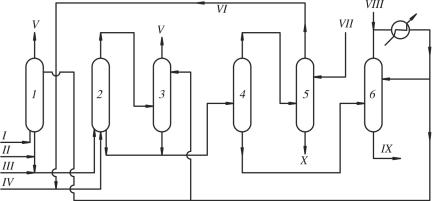
Подробные исследования системы Со2(СО)8 – пиридин (γ-пиколин) позволили разработать промышленный процесс, протекающий в значительно более мягких условиях (температура 150–170 °С, давление 7,5–20 МПа, растворитель ацетон), чем процесс фирмы BASF. Технологическая схема промышленного синтеза пропионовой кислоты по способу фирмы Hulls показана на рис. 11.5.
Катализатор готовят из компонентов в отдельном аппарате и подают в реактор одновременно с олефином, СО и водой (спиртом). Продукты реакции затем поступают в реактор окисления, где получаютокисленнуюформу катализаторного комплекса, которая легко
221

Рис. 11.5. Принципиальная технологическая схема производства жирных кислот карбонилированием олефинов по способу фирмы Hulls: 1 – узел приготовления катализатора; 2 – реактор синтеза; 3 – реактор для окисления катализатора; 4 – колонна отделения воды; 5 – сепаратор; 6–8 – ректификационные колонны. Потоки: I – пиридин (γ-пиколин); II – соль Со; III – СО + Н2; IV – катализатор; V – СО; VI – олефин; VII – вода; VIII – воздух;
IX – кислота; X – кубовыйостаток
отделяется от продуктов реакции в сепараторе и возвращается в цикл. Жидкие вещества после отделения катализатора поступают в узел ректификации, где выделяются пиридин (возвращается на приготовление катализатора), олефин (возвращается в реактор синтеза) и целевой продукт. Конверсия этилена достигает 98 мас. %, селективность образования пропионовой кислоты выше 99 %.
11.3. ПРОИЗВОДСТВО АКРИЛОВОЙ КИСЛОТЫ
Основным промышленным способом получения акриловой кислоты является карбонилирование ацетилена в присутствии никелевого катализатора. По технологии высокого давления фирмы BASF процесс осуществляют при температуре 180–205 °С и давлении 4–8 МПа
222
с использованием в качестве катализатора NiBr2 – галогенида меди в тетрагидрофуране (рис. 11.6). Акриловая кислота получается с выходом 90 % на С2Н2 и 85 % на СО. Годовая мощность установок по технологии высокого давления составляет 132 тыс. т/год (фирма
BASF) и 30 тыс. т/год (фирма Dow-Badische, США).
Рис. 11.6. Принципиальная технологическая схема получения акриловой кислоты карбонилированием ацетилена по способу фирмы BASF: 1 – сатуратор; 2 – реактор; 3 – скруббер; 4 – дегазатор; 5 – абсорбер; 6 – ректификационная колонна. Потоки: I – ацетилен; II – вода; III – катализатор; IV – СО; V – абгаз; VI – газ рецикла; VII – свежая вода; VIII – тетрагидрофуран;
IX – акриловаякислота; X – промывочнаявода
Фирмы Rohm and Haas и Toa Gosei разработали процесс, представляющий комбинацию стехиометрического и каталитического способов, что позволяет работать при низком давлении. По этой технологии акриловую кислоту получают при атмосферном давлении и температуре 30–50 °С с выходом 80–90 % на четырех заводах в США (фирма Rohm and Haas) мощностью 140 тыс. т/год и на заводе в Японии (фирма Toa Gosеi) мощностью 36 тыс. т/год.
223
В последние годы фирма Union Oil (Калифорния) разработала новый перспективный процесс получения акриловой кислоты окислительным карбонилированием этилена по реакции
СН2=СН2 + СО + ½ O 2 → СН2=СНСООН |
(11.4) |
Процесс проводят при температуре 135–150 °С, давлении СО 7–8 МПа и соотношении СО : С2Н4 = 1 : 1 в среде ледяной уксусной кислоты с добавкой до 20 % уксусного ангидрида, 0,1 % PdCl2
и 0,5 % смеси LiCl, LiOCOCH3 и СuС12.
Глава 12
НЕКОТОРЫЕ НОВЫЕ НАПРАВЛЕНИЯ РАЗВИТИЯ ПРОЦЕССА ОКСОСИНТЕЗА
За годы, прошедшие после открытия реакции гидроформилирования и создания первых промышленных установок оксосинтеза, в этой области развивались не только пути усовершенствования оксопроцесса, но и некоторые реакции, родственные гидроформилированию. Наибольший практический интерес представляла разработка процессов на основе реакций:
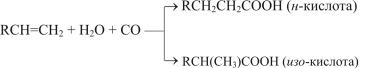
– гидрокарбоксилирования (получения кислот):
RCH=CH2 + CO + H2O → RCH2CH2COOH; |
(12.1) |
– гидроалкоксикарбонилирования (получения сложных эфиров):
RCH=CH2 + CO + R′OH → RCH2CH2COOR′; |
(12.2) |
– карбонилирования спиртов: |
|
RCH2OH + CO → RCH2COOH |
(12.3) |
Как видно из приведенных реакций, донорами водорода в них (вместомолекулярноговодорода) являютсясоответственноводаиспирты.
Эти реакции открывают прекрасные перспективы для получения на их основе широкого ассортимента кислот и эфиров.
12.1. ПОЛУЧЕНИЕ ВЫСШИХ СИНТЕТИЧЕСКИХ ЖИРНЫХ МОНОКАРБОНОВЫХ КИСЛОТ Н-СТРОЕНИЯ НА БАЗЕ РЕАКЦИИ ГИДРОКАРБОКСИЛИРОВАНИЯ
Высшие жирные кислоты н-строения находят широкое использование в производстве синтетических моющих средств бытового
итехнического назначения, поверхностно-активных веществ, автошин
ирезино-технических изделий, смазок, моторных масел, ингибито-
225
ров коррозии, синтетического каучука, а также в кожевенной, лакокрасочной промышленности и других областях.
В промышленном масштабе используют два вида кислот – нату-
ральные и синтетические.
До 90 % вырабатываемых в мире высших кислот н-строения базируется на использовании в качестве сырья растительных масел и натуральных жиров, и пока лишь 10 % – на переработке сырья нефтехимического происхождения. Ежегодно на выработку высших жирных кислот расходуется до 8 млн т натуральных масел и жиров (талового, кокосового, кашалотового и др.).
Ограниченность природных ресурсов, резкое колебание цен на них, а также наметившийся устойчивый дефицит в естественных маслах и жирах не позволяют надеяться на расширение масштабов производства натуральных кислот. Ориентирование потребителей только на натуральные кислоты становится ненадежным. К тому же население Земли постоянно увеличивается. По этим причинам интерес к проблеме синтетических кислот зародился еще в XIX в.
Начало истории развития процессов получения синтетических жирных кислот связано с окислением нефтяных парафинов и относится к 3-й четверти XIX в.
Трудности в обеспечении жирами Германии и ее союзников во время Первой мировой войны послужили толчком для практической реализации методов окисления олефинов. В 1916 г. фирмой «Фанто» была введена в действие первая промышленная установка по производству синтетических жирных кислот. К концу Второй мировой войны в Германии действовало четыре крупных установки по окислению парафинов суммарной мощностью по сырью 110 тыс. т/год. Однако после войны неблагоприятная конъюнктура в ценах на парафин, действовавших в странах Западной Европы, привела к консервации этих установок.
В настоящее время промышленные установки по производству синтетических жирных кислот на базе нефтяных парафинов действуют в Германии, Румынии, Польше, Китае, России и других странах.
Процесс окисления парафинов осуществляется кислородом воздуха при температуре 12–160 °С с использованием в качестве катализато-
226
ра смеси солей марганца и натрия в соотношении Mn : Na = 1 : (1–10). Действующие производства по получению синтетических жирных кислот окислением парафинов имеют ряд существенных недостатков: низкая селективность на стадии окисления (~ 50 %); образование в процессе большого количества сточных вод (до 8 м3 на тонну кислот) и широкойгаммы кислот отС1 доС30; низкоекачество получаемых кислот.
Предпринимаемые усилия исследований по усовершенствованию технологии окисления парафинов не дали ощутимых результатов.
И сегодня действующие производства получения синтетических жирных кислот окисления парафинов имеют ряд существенных недостатков:
–низкое качество кислот, обусловленное присутствием в них различных примесей – неомыляемых, изо-, кето-, окси-, циклопарафиновых кислот;
–образование в процессе большого количества сточных вод – 8 м3 на тонну кислот;
–образование в ходе синтеза широкой гаммы кислот от С1 до С30;
–низкая селективность на стадии окисления парафинов (~ 50 %). Таким образом, острый дефицит в природном сырье, с одной
стороны, и отсутствие эффективной технологии получения синтетических жирных кислот – с другой, ставили вопрос о поиске и разработке новых промышленно-перспективных методов производства высших синтетических кислот н-строения, которые могли бы полностью заменить в технических целях натуральные кислоты.
Фирмами Rurchemi и Host (ФРГ), «Ай-Си-Ай» (Англия), Celenese (США) и другими был разработан и реализован процесс производства синтетических жирных кислот окислением альдегидов оксосинтеза.
Процесс жидкофазного окисления альдегидов проводят при температуре 50–70 °С в присутствии в качестве катализатора солей металлов переменной валентности – Co, Ni, Cr, Mo, V, W, Sn и др.
По сравнению с окислением парафинов, этот процесс является более эффективным. Однако он имеет серьезные недостатки:
– наряду с кислотами н-строения образуется значительное количество (до 45 %) кислот с разветвленной структурой;
227
–при окислении альдегидов С12 и выше заметно возрастает выход побочных продуктов (до 15 %), что приводит к резкому снижению качества кислот. Процесс экономически эффективен лишь для синтеза кислот с числом атомов углерода не выше С11;
–в процессе в качестве сырья используются дорогостоящие оксоальдегиды.
Несомненно наиболее перспективным представляется одностадийный процесс получения синтетических жирных кислот на основе реакции гидрокарбоксилирования:
(12.4)
(12.5)
Впромышленном масштабе эта реакция реализована для получения пропионовой кислоты из этилена с использованием в качестве
катализатора системы [HNi(CO)4 + HJ].
Сведения по гидрокарбоксилированию высших олефинов отражены лишь в ряде патентов, которые посвящены исследованию этой реакции вприсутствии палладия, платины, родия, иридия, осмия и рутения.
Исследование реакции гидрокарбоксилирования высших олефинов представляло как научный, так и прикладной интерес – в плане разработки промышленной технологии получения высших синтетических жирных кислот.
Втечение 1970–80-х гг. во ВНИИНефтехиме были впервые выполнены всесторонние и детальные исследования реакции гидрокарбоксилирования.
Поскольку реакция гидрокарбоксилирования родственная реакции гидроформилирования, то, естественно, мы сосредоточили свое внимание при ее изучении на кобальтовых катализаторах (как в оксосинтезе) в отличие от зарубежных исследований.
Входе проверки различных Со-содержащих катализаторов:
НСо(СО)4, Со-трифенилфосфана, различных Со-пиридиновых комплексов – было установлено, что наиболее эффективным катализатором является комплекс [Co(Py)6] [Co(CO)4]2.
228
Да, это тот самый комплекс, который был активен при гидроформилировании высших олефинов с «сильно» разветвленной цепью (когда рассматривали процесс получения изодеканола).
В ходе исследований кинематической закономерности реакции гидрокарбоксилирования на примере гексена-1 было изучено влияние температуры, добавления окиси углерода и водорода, концентрации катализатора, соотношения олефин : вода и растворителя.
Поскольку в качестве исходных реагентов в реакции гидрокарбоксилирования участвует водная и органическая фазы, то существенное влияние на протекание должен был оказывать растворитель. Экспериментально было установлено, что ассортимент растворителей, способных гомогенизировать (сделать гомогенной) реагирующие компоненты, в условиях реакции довольно ограничен. Это пиридин, циклогексанон, ацетон, диоксан, тетрагидрофуран и этиловый эфир уксусной кислоты. Было установлено, что наибольшие скорости достигаются в пиридине, циклогексаноне и ацетоне (табл. 12.1).
При исследовании влияния температуры было установлено, что наиболее высокие выходы кислот (~ 90 %) достигаются в интервале температур 140–150 °С. При более высоких температурах наблюдается снижение селективности реакции.
Таблица 1 2 . 1
Константы скорости реакции при использовании различных растворителей
№ |
Растворитель |
Константа скорости |
п/п |
К 10–2, мин–1 |
|
1 |
Пиридин |
1,57 |
2 |
Циклогексанон |
1,45 |
3 |
Ацетон |
1,31 |
4 |
Диоксан |
0,74 |
5 |
Тетрагидрофуран |
0,57 |
6 |
Этиловый эфир уксусной кислоты |
0,51 |
Давление окиси углерода также оказывает существенное влияние. Так, при повышенном РСО от 7,5 до 200 МПа скорость реакции увеличивается почти в 3 раза.
229

Дальнейшее повышение давления РСО приводит к снижению селективности процесса за счет протекания реакции образования альдегидов:
RCH=CH2 + 2CO + H2O → RCH2CH2CHO + CO2. |
(12.6) |
При исследовании реакции концентрации катализатора в интервале 0,2–1,2 мас. % по Со был установлен 1-й порядок образования кислот по катализатору. На селективность реакции концентрация катализатора практически не оказывала влияния.
Значения некоторых констант скоростей реакции образования кислот и альдегидов при температуре 140 °С и давлении СО 12,5 МПа приведены в табл. 12.2.
Таблица 1 2 . 2
Зависимость скорости образования кислот и альдегидов от концентрации катализатора
Концентрация катализатора, вес. % |
Константы скорости |
||
образования К 10–2, мин–1 |
|||
(в расчете на металлический кобальт) |
|
|
|
кислот |
альдегидов |
||
|
|||
0,25 |
0,44 |
0,03 |
|
0,50 |
0,90 |
0,06 |
|
1,19 |
1,30 |
0,09 |
|
Технический интерес представляла оценка оптимального избытка воды, подаваемой на реакцию. Было установлено, что оптимальные скорость и селективность реакции достигаются при мольном соотношении вода : олефин, равном 2 : 4; температура 140 °С, давле-
ние СО 12,5 МПа, ССО2(СО)4 = 1,2 мас. % (табл. 12.3).
Небольшие добавки водорода (0,1–0,3 %) приводили к ускорению реакции гидрокарбоксилирования в 1,5–2 раза. При повышении давления РН2 выше 0,3 об. % селективность реакции гидрокарбоксилирования заметно снижалась за счет одновременного протекания реакции гидроформилирования. Так, при РН2 = 1 об. % селективность реакции гидрокарбоксилирования составляла всего 10 %.
230