

5.2 Инсектицидные органические производные кислот фосфора
В пятидесятые годы прошлого века производные кислот фосфора с инсектицидной активностью заменили в арсенале средств защиты растений хлорорганические препараты – ДДТ, гексахлоран, полициклические хлорпроизводные, применение которых создавало серьёзные проблемы для окружающей среды из-за их персистентности, острой и хронической токсичности. В отличие от хлорорганических соединений фосфорорганические достаточно быстро разлагаются в почве и в водных средах с образованием безопасных веществ.
Отправной точкой для разработки этой группы биологически активных веществ стало обнаружение инсектицидной активности у фторангидрида метансульфокислоты CH3SO2F. Это жидкое соединение с высоким давлением паров и сейчас находит ограниченное применение в качестве фумиганта. Затем были получены и фторангидриды различных амидов, эфиров и амидоэфиров кислот фосфора с очень высокой токсичностью для теплокровных. В соответствии с этим дальнейшие исследования в этой области проводились уже с целью получения нового класса отравляющих веществ. Полученные в ходе работ над новыми токсикантами соединения проявляли также хорошие инсектицидные свойства. После окончания Второй Мировой войны работы в области производных кислот фосфора были продолжены в том числе и с целью получения безопасных для теплокровных соединений, которые можно было использовать для борьбы с насекомыми вредителями.
Интенсивные исследования способов получения и инсектицидной активности избирательных по отношению к насекомым производных кислот фосфора, проводившиеся до начала 80‑х гг. прошлого века, завершились получением около 70000 новых соединений. Возможность варьирования четырех заместителей у центрального атома фосфора делает производные фосфорной и тиофосфорных кислот неисчерпаемым источником новых биологически активных веществ. У полученных органических производных кислот фосфора изучалась зависимость инсектицидной активности и токсичности от их строения. Накопленный опыт был положен в основу отбора препаратов с оптимальными токсикологическими характеристиками для организации их производства. К 1970 году поступавшие на рынок инсектицидные препараты включали более 200 различных активных начал на основе кислот фосфора. Однако появление рас вредителей с резистентностью к эфирам и амидам кислот фосфора лишило перспектив дальнейшие синтетические исследования в этой области. Тем не менее выпуск разработанных более пятидесяти лет назад инсектицидов, называемых чаще всего фосфорорганическими, не прекращается до настоящего времени. Новые способы борьбы с членистоногими позволяют использовать их даже против резистентных рас вредителей, несмотря на то, что чувствительность насекомых к этим препаратам значительно снизилась.
Как и у рассмотренных выше хлорорганических препаратов, механизм биологической активности производных кислот фосфора основан на нарушении передачи нервных импульсов. Функционирование нервной системы в организмах теплокровных изучено достаточно хорошо. Проходящий по отростку нейрона (аксону) в виде волны изменения поляризации мембраны аксона слабый сигнал электрической природы реализуется в синапсе, т.е. в точке контакта аксона и иннервируемой клетки. Достигшее пресинаптической мембраны изменение её полярности через опосредованный ионами кальция механизм вызывает выделение в синаптическую щель, разделяющую пресинаптическую и постсинаптическую мембраны, химического вещества – нейромедиатора, вызывающего через соответствующие рецепторы активацию или торможение в иннервируемых клетках. Основными нейромедиаторами возбуждения у теплокровных являются ацетилхолин и норадреналин. Выполнивший свою функцию ацетилхолин инактивируется в результате катализируемого холинэстеразой гидролиза на холин и уксусную кислоту. В организмах теплокровных снижение активности этого фермента приводит к накоплению в нервных окончаниях эндогенного ацетилхолина, вызывающего непрекращающуюся активацию соответствующих биохимических систем. Следствием этого становится исчерпание энергетических ресурсов клеток и их гибель.
Нервная система насекомых и особенности поражения её ингибиторами гидролаз изучены гораздо хуже. Основу нервной системы насекомых составляет серия ганглий, которые мало похожи на периферические узлы ганглий млекопитающих. У насекомых эти образования выполняют роль центральной нервной системы. От проникновения чужеродных веществ они защищены сплошной мембраной, выполняющей роль гематоэнцефалического барьера теплокровных. Иннервация внутренних органов и органов дыхания у насекомых не имеет обратной связи. Передача нервного импульса по аксону протекает за счёт концентрационных градиентов натрия и калия по разные стороны мембраны, но у растительноядных насекомых гемолимфа содержит больше ионов калия, чем натрия и поэтому нервные окончания у них устроены иначе, чем у насекомых хищников.
В восьмидесяты годы ХХ века было известно около двадцати нейропептидов насекомых. В последние годы успехи микроаналитических методик, используемых для изучения пептидов, позволили обнаружить в организмах членистоногих несколько сот различных нейропептидов, участвующих в метаболизме и в миотропных процессах. Один из них – пентапептид проктолин – в концентрациях около 10-10 моль/л вызывает сильное возбуждение мышечных тканей и внутренних органов насекомых. Структура проктолина установлена – это аргинил-тирозинил-лейцил-пролил-треонин. Инактивация выполнивших медиаторную функцию нейропептидов может осуществляться гидролитическим путём, как и инактивация ацетилхолина. Расщепление амидных связей в пептидах проводят протеолитические ферменты, активные центры которых подобны эстеразным. Так, например, активный центр одной из наиболее изученных протеаз альфа-химотрипсина составляют включённые в пептидную цепь фрагменты серина, гистидина, аспарагина и ещё одного остатка гистидина. Эти же структурные элементы присутствуют и в активных центрах зстераз. У эстераз, как и у протеаз, каталитическая активность обусловлена переносом ацильного остатка сложноэфирной или пептидной связи на гидроксильную группу серинового фрагмента в активном центре фермента. Во всяком случае диизопропилфторфосфат одинаково необратимо ингибирует как альфа-химотрипсин, так и холинэстеразу.
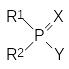
Анализ экспериментального материала, полученного в процессе изучения зависимости токсичности от строения фосфорорганических ингибиторов холинэстеразы, позволил основоположнику этого направления в химии биологически активных веществ немецкому учёному Г.Шрадеру (Gerhard Schrader, 1903-1990) предложить общую формулу для производных кислот фосфора с антихолинэстеразной активностью, известную как формула Шрадера:
 ,
,
где остатки R1 и R2 независимо один от другого означают алкильные, арильные группы, алкоксигруппы, арилоксигруппы, алкиламидные и ариламидные группы, алкилмеркаптогруппы, Х означает атом кислорода или серы, а остаток Y означает уходящую группу, обеспечивающую перенос фосфорильного структурного элемента на нуклеофильную функиональную группу в каталитическом центре холинэстеразы. При этом в роли заместителя Y должны выступать остатки слабых кислот, например, синильной, фтористоводородной кислоты, меркаптана, остаток фосфорной кислоты, также это может быть замещенная электроноакцепторными заместителями арилоксигруппа, енольный остаток и др. Конечно, более эффективными уходящими группами являются, например, атомы галогенов или остатки других сильных кислот, но фосфорилирующая активность, например, хлорангидридов оказывается настолько высокой, что они просто не могут попасть в защищенные глиальными клетками синапсы центральной и периферической нервной системы, где локализована участвующая в передаче нервного импульса холинэстераза. При поступлении в организм такие ангидриды прореагируют с водой или с другими менее важными нуклеофильными группами в составе биомолекул.
Представленная формулой Шрадера рабочая гипотеза достаточно долго определяла направление поиска новых инсектицидов. По мере накопления экспериментальных данных о фосфорилирующей способности соединений, которые не вписываются в рамки этой формулы, она была дополнена. В частности, антихолинэстеразной активностью обладают также производные кислот фосфора с функциональными группами P‑X‑Y‑Z, где Z представляет собой структурный элемент, который может принимать электроны P‑X‑связи. В роли X, Y, и Z выступают обычно атомы H, C, N, O, S или галогены. Связь P–X не должна усиливаться pπ‑dπ‑сопряжением. Необходимо также, чтобы группа Z легко поляризовалась или становилась электроотрицательной в результате метаболического окисления.
Связь P–X бывает ослаблена в тех случаях, когда атом Y находится в sp2‑гибридизованном состоянии. Примером таких инсектицидов могут служить енольные эфиры фосфорной кислоты – дихлофос, Гардона и другие. Если между атомами X, Y и Z имеются только простые связи, то эффективное фосфорилирование возможно по схеме:
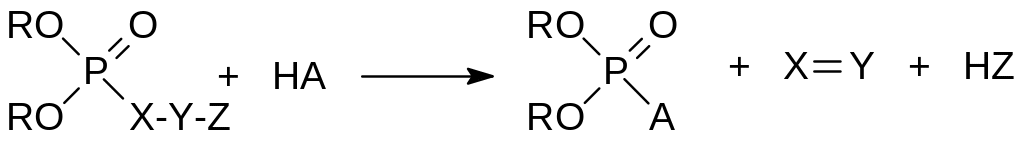
В качестве примера можно привести перенос диметоксифосфорильной группы в составе кислородного аналога инсектицида азинфосметила на субстрат НА в соответствии с уравнением:
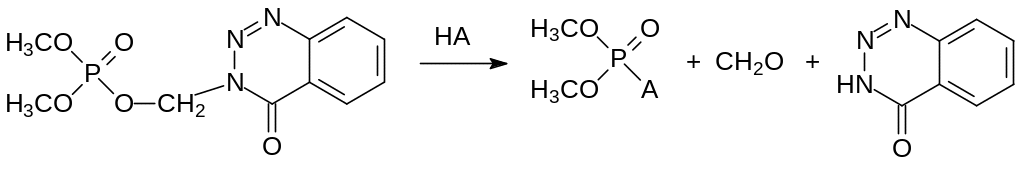
Формула Шрадера и представленное здесь правило представляют собой хорошее руководство для первичной оценки фосфорилирующей способности и токсичности органических производных кислот фосфора по их структурной формуле. Тем не менее в них не учитываются все возможности так называемого «летального синтеза», когда не проявляющее физиологической активности в модельных опытах вещество под действием неспецифических оксидаз и других ферментов в живых системах превращается в высокотоксичное. В соответствии с этим различия в метаболических превращениях ксенобиотиков в клетках теплокровных и членистоногих может быть использовано для получения селективных инсектицидов.
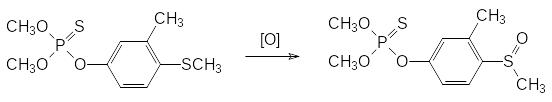
В качестве примера «летального синтеза» можно привести метаболическое окисление атома серы в молекуле с сульфидной функциональной группой. Так, фенольная структурная единица действующего начала препарата Лебайцид (О,О-диметил-О-(3-метил-4-метилмеркаптофенил)тиофосфат) включает только электронодонорные заместители, но, тем не менее, это вещество проявляет высокую инсектицидную активность с токсичностью для крыс около 230 мг/кг. Если принять во внимание, что аналогичное соединение с метоксигруппой вместо метилмеркаптогруппы значительно менее токсично, то остается только предположить, что оксигеназы, предназначенные для метаболического превращения поступающих в организм ксенобиотиков, переводят входящую в состав этого соединения электронодонорную метилмеркаптогруппу в электроноакцепторную метилсульфоксидную группу и это превращение представляет собой один из примеров летального синтеза:
Другой механизм летального синтеза отвечает за антихолинэстеразную активность диметилового эфира 1-гидрокси-2,2,2-трихлорэтилфосфоновой кислоты с ЛД50 для крыс 560 мг/кг. Это никак не соответствующее формуле Шрадера соединение, известное под названием хлорофос, диптерекс и др., в присутствии даже слабых оснований претерпевает фосфонат-фосфатную перегруппировку с отщеплением молекулы хлористого водорода:
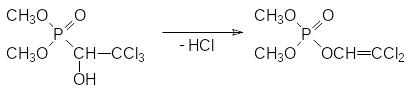
В результате дегидрохлорирования фосфонат с фосфор-углеродной связью превращается в фосфорилированный енол, который уже может выступать в качестве фосфорилирующего средства, поскольку в его составе появляется уходящая группа, соответствующая остатку слабой кислоты. В данном случае речь идет о енольной форме дихлоруксусного альдегида.
Изучение механизма действия соединений с фосфорилирующей способностью показало, что они обратимо или необратимо блокируют ферменты, относящие к классу гидролаз. При этом основной токсический эффект у теплокровных и членистоногих проявляется в результате ингибирования холинэстеразы (ХЭ) – фермента, катализирующего гидролитическое разложение передающего сигнал возбуждения нейромедиатора ацетилхолина на уксусную кислоту и холин:
![]()
Важной особенностью фосфорорганических токсикантов и серьезным недостатком многих фосфорорганических инсектицидов с антихолинэстеразным механизмом действия является их хроническая токсичность, связанная с необратимостью вызванного ими ингибирования холинэстеразы. Этот фермент относится к сериновым гидролазам, в активном центре которых находится гидроксиметильная группа включённой в пептидную цепь белка аминокислоты серина. Катализируемое холинэстеразой гидролитическое разложение ацетилхолина начинается с переноса ацетильной группы молекулы ацетилхолина на гидроксильную группу серинового фрагмента белка при катализе основанием, представленным имидазольным структурным элементом аминокислоты гистидина в пептидной цепочке эстеразы. Затем ацетильная группа отщепляется гидролитическим путём при участии уже другого имидазольного цикла и фермент может снова ацетилироваться, разлагая следующую молекулу ацетилхолина. Однако при ацилировании холинэстеразы фосфорорганическими соединениями с эфирными группами у атома фосфора появляется возможность гидролиза не только по эфирной связи, соединяющей фосфорный остаток с молекулой фермента (обратимое ингибирование), но и по другим эфирным связям у атома фосфора. В результате этого происходит необратимая блокировка холинэстеразы, поскольку атака молекулы воды по фосфорильной группе в анионной форме оказывается невозможной:
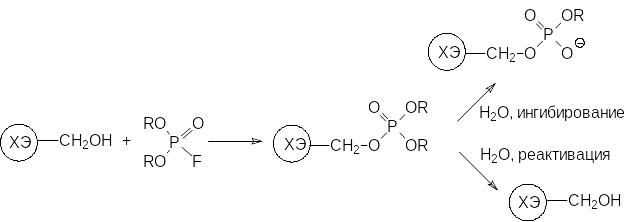
В синапсах биосинтез новых молекул холинэстеразы вместо заблокированных идет очень медленно, и после воздействия сублетальных доз антихолинэстеразных ядов содержание этого фермента уже никогда не выходит на прежний уровень, а его недостаток компенсируется организмом другими путями.
Для лечения острых отравлений соединений с антихолинэстеразной активностью были разработаны антидотные композиции, в состав которых наряду с антагонистами ацетилхолина, обратимыми ингибиторами холинэстеразы и успокаивающими средствами входят различные производные гидроксиламина. При этом антидотный состав должен быть введён в виде инъекции в течение нескольких минут после появления первых признаков отравления для того, чтобы предотвратить необратимое ингибирование основного пула холинэстеразы. Переход фосфорилированной холинэстеразы в состояние, которое не поддается регенерации, часто называют «старением» ингибированной холинэстеразы. Одним из соединений, нейтрализующих попавший в организм токсикант, может быть, например, гидроксамовая кислота на основе никотиновой кислоты. Она взаимодействует с поступившим в организм антихолинэстеразным ядом, нейтрализуя его до того, как он прореагирует с холинэстеразой:
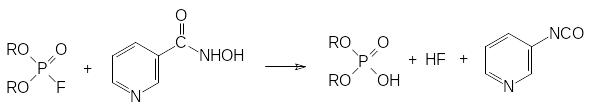
Промежуточным продуктом в этом превращении является фосфорилированная по гидроксильной группе гидроксамовая кислота, которая по реакции Лоссена разлагается на диалкилфосфат и соответствующий изоцианат, гидролизующийся водой с образованием нестойкой карбаминовой кислоты.
Еще одна группа средств для нейтрализации таких ядов представлена монооксимами α-дикарбонильных соединений. Так, например, монооксим диацетила, образующийся при нитрозировании метилэтилкетона, реагирует с диалкилфторфосфатом по схеме:
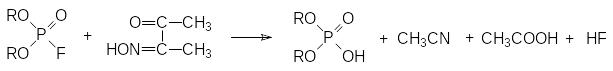
Промежуточным продуктом в этом случае является фосфорилированный по гидроксильной группе оксим, разлагающийся водой с образованием диалкилфосфорной кислоты, уксусной кислоты и ацетонитрила.
Ещё одна группа соединений предназначена для реактивации фосфорилированных молекул холинэстеразы, алкоксифосфорильная группа которых ещё не перешла в результате гидролиза в ионную форму. Такими антидотами являются оксимы альдегидов на основе пиридиниевых солей. Реакция оксима с фосфорилированной холинэстеразой протекает по схеме:
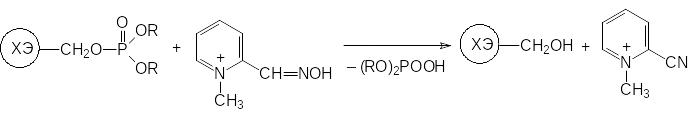
В соответствии с представленной схемой превращений соль оксиминометилзамещенного метилпиридиния (2-ПАМ), получаемая нитрозированием соли N-метил-α-пиколиния, реактивирует фосфорилированную холинэстеразу и превращается в соответствующий нитрил. При этом роль четвертичного атома азота в молекуле 2-ПАМ состоит в фиксации молекулы антидота в активном центре фермента в результате взаимодействия с анионным сайтом, предназначенным для связывания с триметиламмонийной группой в молекуле ацетилхолина.
Многие О-фосфорилированные производные гидроксиламина соответствуют формуле Шрадера и, в согласии с этим, они должны быть антихолинэстеразными ядами. В качестве примера таких соединений можно назвать инсектицид фоксим и один из токсинов сине-зеленых водорослей анатоксин:
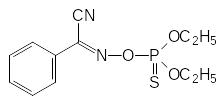
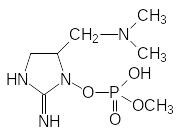
Фоксим Анатоксин а(s)
Из этого следует, что представленные выше гидроксамовые кислоты, оксимы α‑дикарбонильных соединений и оксимы альдегидов можно использовать в качестве антидотов только потому, что продукты их фосфорилирования разлагаются на малотоксичные соединения сразу после образования.
В дополнение к этому можно отметить, что кроме синаптической холинэстеразы в организме теплокровных есть еще эритроцитарная холинэстераза, связанная с мембранами эритроцитов, а также сывороточная холинэстераза, растворенная в плазме крови. Они предназначены для гидролиза ацетилхолина и других продуктов ацилирования холина, образующихся в организме в результате взаимодействия с холином молекул ацетилкофермента А или кофермента А, ацилированного остатками других кислот:
![]()
Реакционная способность образующихся из кофермента А тиоэфиров карбоновых кислот достаточно высока для того, чтобы ацилировать холин без участия катализатора (такие реакции, протекающие в биологических средах без ферментативного катализа, называют параметаболическими). Образующиеся при этом ацильные производные холина могут взаимодействовать с холинорецепторами, создавая ненужное фоновое возбуждение, но они практически не гидролизуются высокоизбирательной холинэстеразой. В соответствии с этим содержание в жидких средах организма ацетилхолина и его аналогов со свойствами агонистов должно быть сведено к минимуму. Именно для этого и предназначены несинаптические эритроцитарная и сывороточная холинэстеразы. Сывороточная холинэстераза, в отличие от истинной или эритроцитарной, неизбирательна, она имеет более низкую молекулярную массу и поэтому растворима в плазме крови. Её субстратами являются эфиры холина и различных эндогенных карбоновых кислот. Сывороточная холинэстераза показывает небольшую избирательность по бутирилхолину, из‑за чего её иногда называют бутирилхолинэстеразой или ложной холинэстеразой. Истинная холинэстераза представляет собой тетрамер с молекулярной массой около 335 кДа, т.е. она состоит из четырёх субъединиц с молекулярной массой около 80 кДа. Такие высокомолекулярные белки нерастворимы в воде и поэтому они должны быть связаны с клеточными мембранами. В качестве носителя истинной холинэстеразы, предназначенной для гидролиза образующегося вне нейронов параметаболического ацетилхолина, природа выбрала мембрану эритроцитов, циркулирующих с кровью по всему организму.
Многократное поступление в организм человека даже минимальных количеств необратимых ингибиторов холинэстеразы в процессе их производства и при контактах с препаративными формами фосфорорганических инсектицидов приводит к снижению уровня этого фермента в нервных окончаниях. До определенного предела это может компенсироваться организмом, например, за счёт активации обратной сорбции ацетилхолина через постсинаптическую мембрану или путём уменьшения количества ацетилхолина, поступающего при передаче нервного импульса в синаптическую щель. Однако при этом следует принимать во внимание, что у контактировавших с необратимыми ингибиторами холинэстеразы работников из-за пониженного общего уровня холинэстеразы появляется возможность получения отравления дозами, которые у людей с нормальным уровнем фермента могли вызвать лишь незначительные отклонения в состоянии здоровья.
В рамках работ по синтезу и изучению биологической активности производных кислот фосфора, проводившихся под руководством Г.Шрадера в предвоенные и военные годы, было показано, что многие из них оказываются токсичными для насекомых. Так, например, высокую инсектицидную активность показывал диэтил-4‑нитрофенилфосфат (соединение Е‑600, параоксон), токсичность которого даже превосходила токсичность эфироамида цианфосфорной кислоты, производившегося как отравляющее вещество табун. Переход от фосфата Е-600 к тионфосфату (препарат Е-605, тиофос, паратион)
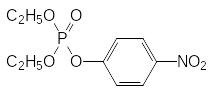 и
и
позволил при сохранении высокой инсектицидной активности значительно понизить токсичность нитрофенилового эфира тиофосфорной кислоты для теплокровных. Дальнейшее снижение токсичности достигалось заменой этильных групп на метильные. Полученный в результате такой модификации структуры параоксона О,О‑диметил-О-4-нитрофенилтиофосфат (ЛД50 25-50 мг/кг) под названием метафос некоторое время использовался в качестве средства для борьбы с насекомыми вредителями, однако достаточно скоро и от него пришлось отказаться из-за большого числа тяжёлых поражений, вызванных неосторожным обращением с инсектицидными составами на его основе, тем более, что вскоре после его внедрения в практику появились новые более безопасные фосфорорганические инсектициды. В ряду разработанных в самом начале этих исследований препаратов можно отметить названный по имени Г.Шрадера препарат Шрадан (Октаметил, октаметилтетрамид пирофосфорной кислоты с ЛД50 9 мг/кг):
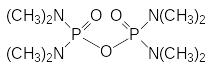
Важной особенностью этого соединения является обратимость фосфорилирования им холинэстеразы и отсутствие у него кожно-резорбтивной активности.
Обратимыми ингибиторами холинэстеразы являются соответствующие формуле Шрадера соединения, у которых заместители у атома фосфора представлены диалкиламидными, алкильными или арильными группами. В частности, кроме представленного выше шрадана таким обратимым ингибитором холинэстеразы является фторангидрид тетраметилдиамида фосфорной кислоты, который может быть получен действием диметиламина и безводного фторида натрия на диметиламид дихлорангидрида фосфорной кислоты:
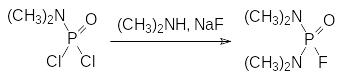
Некоторое время под названием димефокс его использовали в качестве системного инсектицида. Важным преимуществом этого растворимого в воде вещества является также отсутствие у него кожно-резорбтивной активности. При токсичности на крысах при энтеральном введении от 3 до 5 мг/кг он был менее опасен, чем, например, тиофос с токсичностью более 30 мг/кг. Его структурный аналог – бис(изопропиламидо)фторфосфат (мипафокс) – при более низкой токсичности, составлявшей от 25 до 50 мг/кг, оказался даже более опасным в обращении, поскольку замена метильных групп на изопропильные повышала липофильность этого соединения и связанную с этим возможность сорбции через кожу.
Тем не менее, основное направление при разработке новых инсектицидов на основе кислот фосфора было представлено прежде всего поиском соединений с низкой общей токсичностью для теплокровных. Так, например, в ряду ариловых эфиров тиофосфорных кислот серьёзное снижение токсичности при практически полном сохранении инсектицидной активности достигается введением заместителей (атома хлора или метильного остатка) в мета‑положение нитрофенильной группы тионфосфатов. Способ получения этих соединений основан на взаимодействии диметилхлортиофосфата с соответствующими фенолятами.
Для получения метиловых и этиловых эфиров хлорангидридов тиофосфорных кислот проводят взаимодействие тиофосфорилхлорида с метиловым или этиловым спиртом. Взаимодействие этих реагентов в соотношении в пределах от 1 до 3,0-4,5 при охлаждении приводит к замещению только одного атома хлора в молекуле этого хлорангидрида с низкой реакционной способностью, например:

Для замещения на алкоксигруппу ещё одного атома хлора в полученном дихлориде к нему при интенсивном охлаждении прибавляют раствор эквивалентного количества гидроксида натрия в соответствующем спирте:

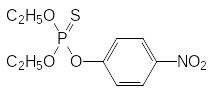
Далее проводят взаимодействие полученного хлорангидрида О,О‑диалкилтиофосфорной кислоты с 3-метил-4-нитрофенолятом натрия:
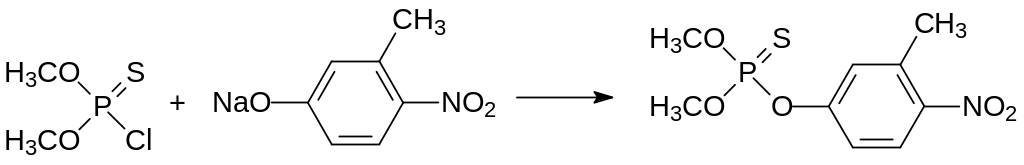
Токсичность получаемого по этой схеме фенитротиона составляет от 250 до 430 мг/кг. Ещё одним препаратом из этой группы является хлортион (ЛД50 около 900 мг/кг), образующийся при фосфорилировании 4‑нитро-3-хлорфенола:
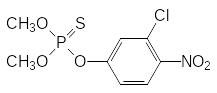
Интересно, что для получения 4-нитро-3-хлорфенола и 3-метил-4-нитрофенола можно использовать нитрование трис(3-хлорфенил)фосфата или трис(м-крезил)фосфата с последующим гидролизом образовавшегося продукта нитрования, например:
![]()
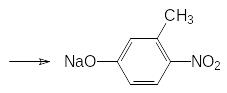
В этом случае стерический фактор в триарилфосфате значительно снижает содержание продуктов орто-замещения в образующихся при нитровании реакционных массах. Тиофосфаты на основе замещенных 2‑нитрофенолов неактивны.
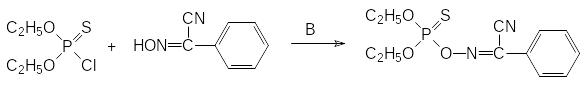
Оптимальным соотношением токсичности и инсектицидной активности характеризуется тиофосфорилированный оксим (фоксим, ЛД50 2000 мг/кг), который получают в результате фосфорилирования хлорангидридом диэтилтиофосфорной кислоты в присутствии основания (В) цианоксима, образующегося при нитрозировании бензилцианида,:
Серьёзный ущерб животноводству наносят оводы, личиночные стадии которых паразитируют в пищеварительном тракте и в теле животных. Готовые к окукливанию личинки оводов мигрируют в подкожные ткани, где и окукливаются, растворив при этом гидролазными ферментами небольшой участок кожи животного для того, чтобы к куколке мог поступать кислород воздуха. Через этот же свищ и выходит вылупившееся насекомое. Понятно, что эти эндопаразиты наносят большой урон животноводству, тем более, что кожа пораженного оводами животного не может быть использована для получения из нее кожевенных изделий. В качестве средства для борьбы с эктопаразитами и системного инсектицида в органиченном объёме используется трихлорметафос-3:
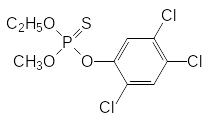
Для борьбы с личинками овода его вводят внутрь в дозах 15-20 мг/кг массы животного в 50%-ном растворе в растительном масле. Токсичность трихлорметафоса-3 составляет около 1100 мг/кг. Его получают фосфорилированием О-метил-О-этилхлортиофосфатом натриевой соли 2,4,5-трихлорфенола, но, как отмечалось выше, масштабное производство этого соединения прекращено из-за присутствия в нём высокотоксичного тетрахлордибенздиоксина.
В настоящее время для борьбы с кожным оводом предлагается использовать растворимый в воде хлорофос (диметиловый эфир 1‑гидрокси-2,2,2-трихлорэтилфосфоновой кислоты, диптерекс, гиподермин-хлорофос) с токсичностью на крысах около 400 мг/кг, который получают по реакции диметилфосфита с хлоралем. В течение многих лет хлорофос был одним из самых популярных инсектицидов для дома и приусадебного хозяйства.
Хлорофос образуется в реакции хлораля с диметиловым эфиром фосфористой кислоты:
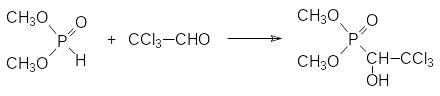
Диметилфосфит в промышленных условиях получают прибавлением трихлорида фосфора к раствору метилового спирта в жидком метилхлориде с температурой кипения -24,2°С. Метилхлорид является побочным продуктом в реакции трихлорида фосфора с метиловым спиртом:
![]()
Такое протекание реакции с превращением промежуточно образующихся в реакции трихлорида фосфора с метиловым спиртом трикоординированных эфиров фосфористой кислоты в диметилфосфит объясняется протонированием атома фосфора в триэфире с элиминированием одной из алкильных групп в виде метилхлорида. Реакция протекает с выделением тепла, для отвода которого используют внешнее охлаждение и испарение метилхлорида, который отмывают от хлористого водорода, конденсируют и возвращают в процесс, отбирая часть материального потока в качестве товарного продукта, используемого, например, в производстве кремнийорганических соединений. В лабораторных условиях диметилфосфит получают прибавлением трихлорида фосфора к охлаждаемому раствору метанола в дихлорметане или в хлороформе.
Превращение хлорофоса в более летучий и более токсичный диметилдихлорвинилфосфат (дихлофос, ДДВФ) было представлено выше (с. ) при обсуждении механизмов «летального синтеза». Понятно, что это превращение, протекающее при действии оснований, может быть реализовано и в крупнотоннажном производстве, однако основной способ получения дихлофоса с токсичностью (ЛД50) для кроликов около 80 мг/кг представлен взаимодействием триметилфосфита с хлоралем по реакции Перкова:
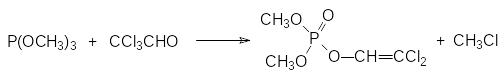
Производство триметилфосфита включает две стадии. Сначала при нагревании проводят взаимодействие трихлорида фосфора и фенола и проводят переэтерификацию образовавшегося трифенилфосфита метанолом, возвращая выделяющийся фенол на стадию получения трифенилфосфита:
![]()
![]()
Трифенилфосфит в отличие от триметилфосфита не реагирует с хлористым водородом даже при нагревании, поскольку фенильная группа не образует карбокатион, а проведение реакции трихлорида фосфора с метанолом в присутствии акцепторов хлористого водорода оказывается более затратным.
Менее токсичное для теплокровных (для крыс ЛД50 430 мг/кг) производное фосфорной кислоты с высокой инсектицидной активностью дибром или налед получают бромированием дихлофоса:
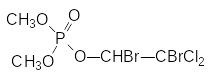
По реакции Перкова были получены и многие другие винилфосфаты, например, по реакции триметилового эфира фосфористой кислоты с галогензамещенными производными ацетоуксусной кислоты или с хлорированными ацетофенонами, но они оказались слишком токсичными. Ещё один способ получения винилфосфатов представлен реакцией соответствующих енолятов натрия с диалкилхлорфосфатами. Так, например, при взаимодействии диметилхлорфосфата с натриевым производным ацетоуксусного эфира образуется диметилфосфат енольной формы ацетоуксусного эфира (фосдрин, мевинфос, ЛД50 3,5 мг/кг, крысы), с высокой инсектицидной активностью:
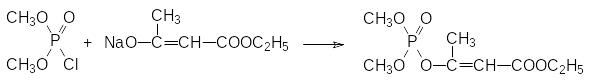
Однако высокая острая токсичность этого соединения не позволила использовать его в широких масштабах. Интересно, что пространственное строение этого соединения решающим образом сказывается на его инсектицидной активности: фосфорилированный Z-изомер на два порядка более активен, чем Е-изомерный фосфат.
Менее токсичен для теплокровных дикротофос, образующийся по аналогичной схеме из диметилхлорфосфата и натриевого производного диметиламида ацетоуксусной кислоты:

Этот инсектицид производится в ограниченных масштабах и в ХХI веке.
В качестве исходных продуктов для получения широкого ряда инсектицидов с невысокой токсичностью для теплокровных используются эфиры дитиофосфорной кислоты, образующиеся из соответствующего спирта и декасульфида фосфора:

Присоединение О,О‑диметилдитиофосфата к диэтиловому эфиру малеиновой кислоты при катализе основаниями приводит к образованию соединения с инсектицидной активностью, известному как карбофос (малатион, ЛД50 500-1500 мг/кг). Его применяют в основном в сельском хозяйстве:
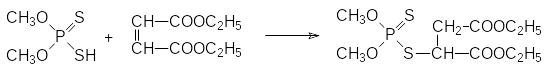
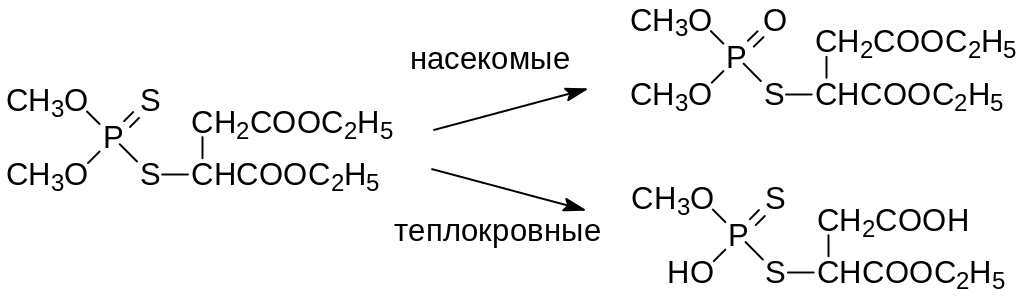
В бытовых условиях производные дитиофосфорных кислот не используются из-за неприятного запаха, источником которого являются образующиеся при их разложении тиольные соединения. Высокая избирательность карбофоса основана на том, что в организме насекомых это малотоксичное соединение окисляется с превращением тиофосфорильной группы в фосфорильную, что приводит к получению вещества с инсектицидной активностью, а в организме теплокровных идёт гидролиз сложноэфирных групп и образуются малотоксичные тионные метаболиты с кислотными функциональными группами:
Малатион можно также получать алкилированием соли диметилдитиофосфорной кислоты диэфиром бромянтарной кислоты, образующимся в результате присоединения бромистого водорода к эфиру малеиновой кислоты. Широкое распространение получили также продукты алкилирования диалкилдитиофосфатов производными альфа-галогензамещенных. карбоновых кислот. Первым соединением этой группы стал ацетион (ЛД50 1050-1100 мг/кг):

Метаболизм ацетиона в насекомых начинается с окисления тионной группы, приводящего к образованию более токсичного тиолфосфата.
Продукт взаимодействия диметилдитиофосфата натрия с эфиром альфа-бромфенилуксусной кислоты – фентоат, цидиал, эрузан (ЛД50 250 мг/кг) – имеет более широкий спектр инсектоакарицидной активности, чем ацетион:
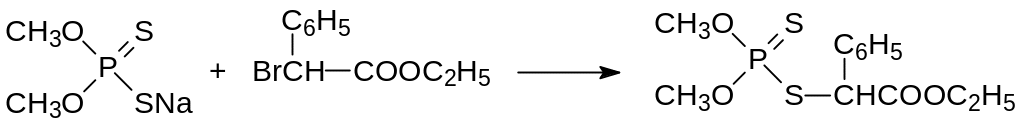
Наиболее известным производным в ряду фосфорилированных тиогликолевых кислот стал O,O-диметил-S-метилкарбамоилметил-дитиофосфат – действующее начало инсектицидов Фосфамид, Рогор, Диметоат (ЛД50 215-267 мг/кг). Основной способ получения этого вещества основан на конденсации диметилдитиофосфата натрия с N‑метилхлорацетамидом:

Известны и другие методы получения этого инсектицида с ярко выраженным системным действием. Фосфамид применяли для борьбы с сосущими насекомыми и паутинным клещиком. В отличие от многих других дитиофосфатов фосфамид лишён кожнорезорбтивной активности и очень быстро распадается на безопасные соединения. Остаточные концентрации фосфамида. в продуктах питания очень низки и выдерживают самый строгий санитарный контроль.
В организмах теплокровных и в растениях фосфамид окисляется, с образованием O,O-диметил-S-метилкарбамоилметилтиофосфата с ЛД50 50 мг/кг. Это вещество предложено использовать в качестве системного инсектицида и акарицида (Ометоат, Фолимат). Для его синтеза предложена оригинальная методика, основанная на взаимодействии с метилизоцианатом продукта фосфорилирования тиогликолевой кислоты диметилхлорфосфатом:
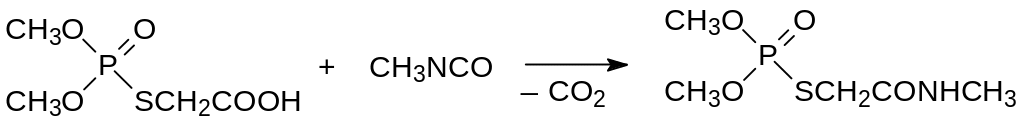
По аналогичной реакции может быть получен и фосфамид.
Представленные выше продукты алкилирования диэфиров дитифосфорных кислот галогенированными производными карбоновых кислот могут рассматриваться также как S-фосфорилированные производные карбоновых кислот с тиольными группами. Сопоставление их структур с естественным субстратом холинэстеразы ацетилхолином показало ещё одно направление поиска новых ингибиторов этого фермента. Структурными аналогами ацетилхолина являются 2‑аминоэтиловые эфиры диалкилтиофосфорных кислот. Первым инсектицидом в ряду аминоалкилтиолфосфатов стал очень токсичный (ЛД50 3 мг/кг) амитон. Он может быть получен по реакции диэтилхлортиофосфата с диэтилэтаноламином с последующей изомеризацией тионфосфата в тиолфосфат по реакции Пищимуки:
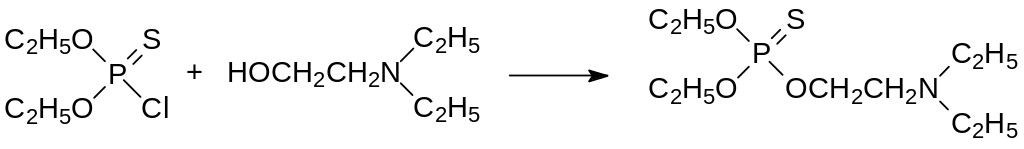

Интересный способ получения амитона основан на взаимодействии натриевой соли диэтилфосфористой кислоты с 2-роданотриэтиламином:
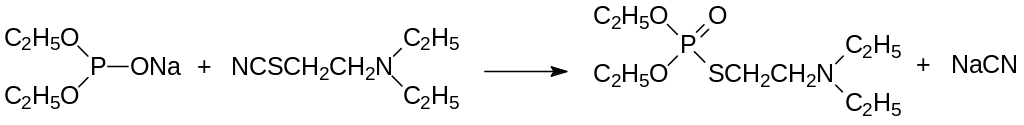
Модифицированием структуры амитона были получены так называемые тиохолиновые эфиры, один из которых производился как боевое отравляющее вещество с обозначением VX. Его пероральная токсичность составляет 0,07 мг/кг – он представляет собой самое токсичное соединение, полученное синтетическим путём.
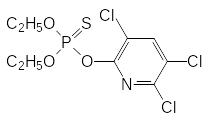
Достаточно безопасные инсектициды были получены на основе азотсодержащих гетероциклических соединений, в составе которых роль связывающегося с анионным центром холинэстеразы основного центра играл атом азота в гетероароматическом цикле. Некоторые из них производятся и ХХI веке, например, хлорпирифос (дурсбан, ЛД50 около 150 мг/кг) и пиримифос-метил (актеллик, камикадзе, ЛД50 около 1400 мг/кг):
 и
и
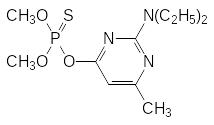
Хлорпирифос применяют для борьбы с москитами и в качестве ларвицида, то есть средства для борьбы с насекомыми в личиночной стадии развития. По ларвицидной активности он в деcятки раз превосходит другие фосфорорганические инсектициды.
Высокой инсектицидной активностью отличается амид О,S-диметил-тиофосфорной кислоты (тамарон, монитор, ЛД50 30 мг/кг), получаемый, например, при метилировании соли эфироамида монотиофосфорной кислоты диметилсульфатом по схеме:
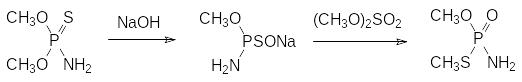
Это соединение может быть также получено в результате термической тион-тиольной изомеризации (реакция Пищимуки) амида диметилового эфира тионфосфорной кислоты в присутствии диметилсульфата:
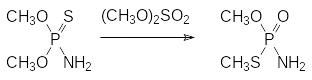
До настоящего времени используется N-ацетильное производное этого соединения (ацефат, ЛД50 около 950 мг/кг). Его получают действием на тамарон ацетангидрида в присутствии каталитического количества концентрированной серной кислоты:
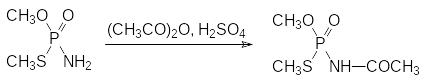
Продолжается производство высокотоксичного контактного и системного инсектицида форат (Тимет, ЛД50 2 мг/кг). Его получают конденсацией О,О‑диэтилдитиофосфорной кислоты с этилмеркаптаном и формальдегидом:
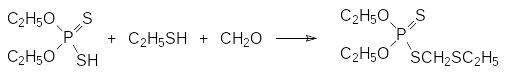
Другой способ получения фората представлен взаимодействием натриевой соли О,О‑диэтилдитиофосфорной кислоты с хлорметилэтилсульфидом, который получают по реакции хлорметилирования действием хлористого водорода и формальдегида на этилмеркаптан:
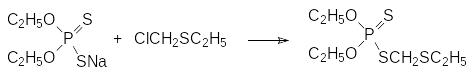
Такие высокотоксичные соединения, как форат, используют преимущественно в качестве почвенных инсектицидов для борьбы с проволочником, многоножками и другими вредителями, повреждающими корневую систему растений и корнеплоды.
Сейчас в производстве инсектицидных препаративных форм находят применение около двадцати действующих веществ на основе кислот фосфора. Для преодоления проявившейся к ним резистентности нормы их расхода надо повышать в несколько раз по сравнению с теми, которые использовались в начальной стадии применения препаратов этого класса, или же применять их в смеси с инсектицидами с другими механизмами действия, используя эффект синергизма. В качестве компонент таких смесевых инсектицидных составов можно использовать неоникотиноиды и карбаматные инсектициды, при этом некоторые из таких смесевых составов демонстрируют даже отрицательную резистентность, т.е. к их действию более чувствительными оказываются резистентные к инсектицидам на основе кислот фосфора расы насекомых.
Важную роль в проявлении инсектицидных свойств играет концентрационный фактор. В соответствии с этим для сохранения норм расхода на единицу поверхности при использовании препаративных форм с высоким содержанием действующего вещества (до 95 масс. %) можно уменьшать размер капель до размеров 100-250 мкм. При этом часто происходит даже экономия пестицида, поскольку концентрированные составы более эффективны, а мелкие капли лучше удерживаются на листьях растений и не стекают с них на почву. Для малообъемного и ультрамалообъемного опрыскивания предназначена специальная аппаратура с ротационным дроблением распыляемого жидкого состава.
В дополнение к изложенному можно привести еще историю практического применения инсектицида фосвел (Лептофос). Это соединение, представляющее собой 4-бром-2,5-дихлорфениловый эфир О‑метилфенилтиофосфоновой кислоты было получено в качестве структурного аналога инсектицида бромофос:

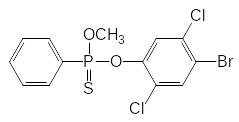
Бромофос Фосвел
Американская компания Velsicol Chemical Corporation организовала производство этого соединения из фенилдихлорфосфина, образующегося при термическом фосфорилировании бензола трихлоридом фосфора. Уже на стадии производства фосвела отмечалось ухудшение состояния здоровья рабочего персонала. Это выражалось в том, что контактировавшие с фосвелом люди теряли ориентацию в пространстве, работоспособность и речевые навыки, из-за чего их называли фосвел-зомби. Исследования механизма такого токсического эффекта показали, что сублетальные дозы фосвела вызывают гибель олигодендроцитов – глиальных клеток, обеспечивающих миелиновую изоляцию аксонов. Демиелинизация аксонов лежит также в основе рассеянного склероза – тяжелейшего аутоиммунного заболевания.
В соответствии с этим инсектицидные средства на основе этого действующего начала не прошли регистрацию в США. Тем не менее компания продолжила производство этого препарата и в течение нескольких лет поставляла его в другие, прежде всего развивающиеся страны. Этот случай демонстрирует, что даже присоединившиеся к программам США по запрету наиболее опасных пестицидов страны третьего мира могут получать от интернациональных корпораций высокоопасные соединения.
В этой связи можно также обратить внимание на нейротоксичность некоторых органических производных кислот фосфора. Впервые она была отмечена на три-о-крезилфосфате (ТОКФ). Хронический нейротоксический эффект ТОКФ впервые проявил себя во время действия с 1920 по 1933 г. "сухого закона" в США, где нелегально распространялся фальсифицированный ром с добавками ТОКФ в качестве имитатора имбирного вкуса. ТОКФ производили для использования в качестве присадки к моторным маслам и в качестве пластификатора для пластмасс. Для человека смертельная доза ТОКФ при поступлении через желудочно-кишечный тракт составляет около 1,0 г/кг, тогда как неоднократное поступление его в организм в дозах от 6 до 7 мг/кг вызывает тяжёлый паралич, в основе которого лежит отмирание длинных аксонов соматической нервной системы.