

Фосген
.docx
Фосген – хлорангидрид угольной кислоты. Продукт многотоннажного синтеза – в 2015 г. 8,5 млн. тонн. Использовался как отравляющее вещество. Его токсическое действие основано на химической модификации структуры альвеолярных белков за счёт ацилирования входящих с их состав аминных групп и азотсодержащих гетероциклов. Мембраны с изменившими третичную структуру белками пропускают плазму крови, что приводит к заполнению альвеолярных пузырьков жидкостью. Это и есть отёк лёгких. Пораженная фосгеном лёгочная ткань не восстанавливается, вместо неё образуется очаг соединительной ткани, который имеет тенденцию к распространению на соседние участки здоровой лёгочной ткани. Со временем следствием этого становится эмфизема и в критических случаях – удушье через много лет после отравления фосгеном.
Фосген – это газ, конденсирующийся в жидкость при температуре около 8 °С. Его получение в промышленности основано на каталитической реакции, протекающей при нормальном давлении и температуре 150-200°С в присутствии активированного угля:
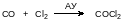
В лабораторных условиях фосген можно получать из четырёххлористого углерода и олеума:
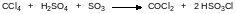
Известен также способ гидролиза ЧХУ с катализом гидратом трихлорида железа или трихлоридом церия:
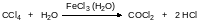
Сейчас появились в продаже генераторы фосгена, представляющие собой небольшие пробирки, в которые загружен трифосген – гексахлордиметилкарбонат – и катализатор его разложения – высококипящий азотсодержащий гетероцикл или фталоцианиновый пигмент. Разложение трифосгена другими катализаторами протекает с низким выходом по фосгену, например:

Скорость выделения фосгена из трифосгена в поступающих в продажу генераторах определяется температурой в области 150°С.
Фосген достаточно медленно гидролизуется водой и парами в воды в воздухе. Скорость и направление его реакции со спиртами зависят от температуры. При охлаждении до 0°С реакция со спиртами идёт преимущественно с образованием моноэфиров хлоругольной кислоты, которые часто называют хлорформиатами:
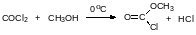
Моноэфиры хлоругольной кислоты используются в пептидном синтезе и в других случаях для активации карбоксильной группы в реакции ацилирования. Из них получают смешанные ангидриды карбоновых кислот и моноэфира угольной кислоты, которые ацилируют нуклеофильные центры с введением ацильной группы и с выделением спирта и диоксида углерода (разложение моноэфира угольной кислоты).
С фенолами фосген не реагирует даже при нагревании. Для получения арилхлорформиатов нужен катализ третичными аминами или нуклеофильный катализ, например, диметилформамидом:
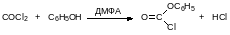
Для получения диалкилкарбонатов фосген пропускают в избыток спирта при нагревании до температуры около 70°С. Ещё один способ получения диалкилкарбонатов основан на окислительном карбонилировании спиртов в присутствии катализаторов на основе одновалентной меди. Так, например, получают используемый в качестве «зелёного» растворителя и реагента диметилкарбонат:
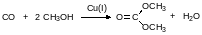
С первичными аминами фосген реагирует с образованием мочевин и карбамоилхлоридов, которые могут разлагаться на изоцианат и хлористый водород. Действие фосгена на первичный амин – это основной способ получения ароматических изоцианатов. В технологическом плане процесс фосгенирования ароматического амина осуществляется в две стадии. Сначала в высококипящем растворителе (хлорбензол) при охлаждении растворяют фосген в количестве, которое превышает стехиометрическое количество из расчета на соответствующий амин. Это нужно для того, чтобы минимизировань образование диарилмочевины, которая с фосгеном не реагирует. После этого при охлаждении к раствору фосгена прибавляют амин и получают реакционную массу, содержащую в основном карбамоилхлорид и гидрохлорид амина:
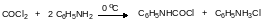
Затем реакционную массу нагревают при пропускании фосгена и доводят её температуру до температуры кипения растворителя. При этом идёт частичная диссоциация соли амина, который фосгенируется в соответствии с представленным выше уравнением и одновременно идёт обратимое разложение карбамоилхлорида на изоцианат и хлористый водород, который уносится парами кипящего растворителя и фосгеном:
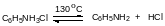
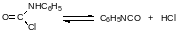
Процесс фосгенирования при повышенной температуре следует проводить при достаточно сильном разбавлении реакционной массы для того, чтобы минимизировать образование бесполезной диарилмочевины за счёт избытка фосгена:

С вторичными аминами в зависимости от соотношения реагентов образуются карбамоилхлорид и тетразамещённая мочевина. Побочный продукт – гидрохлорид амина надо возвращать в процесс или сжигать. В окружающей среде вторичные амины легко превращаются в канцерогенные нитрозамины.
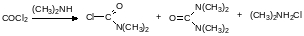
С третичными аминами на холоду фосген образует кристаллические аддукты, которые при нагревании разлагаются на алкилхлорид и карбамоилхлорид, например:
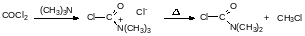
Фосген широко используется в промышленности. Основными потребителями его являются производства поликарбонатов, изоцианатов и прежде всего диизоцианатов, используемых для получения полиуретанов. Исходными продуктами в производстве полиуретанов являются прежде всего 2,4‑толуилендиизоцианат, 4,4´-дифенилметандиизоцианат и гексаметилендиизоцианат:
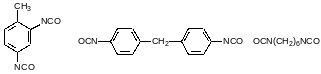
Полиуретаны получают по реакции дизоцианатов с диолами, например, с этиленгликолем:

При получении пенополиуретанов к диольному соединению добавляют воду. Вода реагирует с изоцианатными группами с образованием амина и диоксида углерода, поскольку продукт присоединения воды к изоцианатной группе – это карбаминовая кислота, разлагающаяся на амин и СО2:
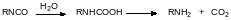
Диоксид углерода вспенивает реакционную массу, а образовавшийся амин реагирует с изоцианатом с образованием замещённой мочевины, сохраняющей способность к полимеризации с диолом:
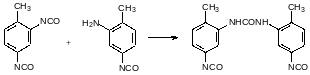
Горение полиуретанов сопровождается их термической деструкцией с образованием очень токсичных изоцианатов. Особенно опасны в этом отношении полиуретаны на основе гексаметилендиамина. Для минимизации рисков, связанных с выделением при пиролизе летучих изоцианатов проводят парциальную полимеризацию изоцианатных групп с образованием, например, триазинтрионовых структурных элементов:
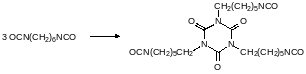
Существуют также бесфосгенные методы синтеза изоцианатов, например, алифатические изоцианаты можно получать алкилированием соответствующими галогенидами солей циановой кислоты. А ароматические изоцианаты можно получать карбонилированием нитропроизводных. Реакция монооксида углерода с нитросоединениями протекает в присутствии родиевых катализаторов. В общем эта реакция должна описываться уравнением, например:
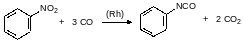
Однако лиганды комплексного родиевого катализатора вызывают полимеризацию изоцианатных групп. Поэтому такой способ получения изоцианатов используют только в тех случаях, когда целевым продуктом является соединение, в которое изоцианат должен был превратиться на следующей стадии. В частности, если изоцианат должен был на следующей стадии превратиться в карбамат, то его синтез карбамоилированием нитросоединения проводят в присутствии соответствующего спирта, например:
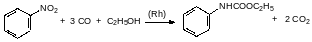
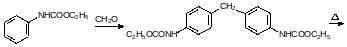
При температуре выше 220°С карбаматы разлагаются на спирт и изоцианат. Если при пиролизе образуется высококипящий изоцианат, то это также может быть использовано для исключения фосгена из синтеза полиуретанов. Так, например, для получения дифенилметандиизоцианата была предложена последовательность превращений, начинавшаяся с О‑этилфенилкарбамата:
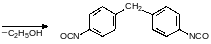
Ещё одна важная группа полимеров на основе угольной кислоты – это поликарбонаты. Сейчас в основном в качестве дигидроксильной компоненты выбирают дифенилолпропан (бисфенол А), который получают конденсацией ацетона с фенолом в присутствии кислот:
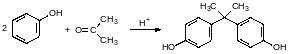
Фенолы реагируют с фосгеном только в присутствии нуклеофильных катализаторов или акцепторов хлористого водорода. В соответствии с этим для производства поликарбонатов проводят реакцию фосгена с растворами бисфенола в щелочах или в присутствии пиридина. Ещё один способ представлен переэтерификацией дифенилкарбоната бисфенолом А в присутствии алкоголята натрия при температуре до 300°С.
Термопластичные поликарбонаты отличаются от других полимеров высокой вязкостью, они не трескаются и не раскалываются. Из них делают остекление для теплиц, большие ёмкости для воды, компакт-диски и многое другое.
Не имеет практического значения бромангидрид угольной кислоты, а дииодангидрид угольной кислоты нестабилен. При взаимодействии фосгена с иодидом натрия продуктами реакции становятся монооксид углерода и иод. Это означает, что образовавшийся в результате обмена атомов галогенов иодфосген сразу разлагается на эти составляющиеся:
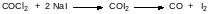
Эта реакция используется для анализа сред на содержание фосгена. Образовавшийся иод титруют тиосульфатом.
Определённый интерес в синтетическом плане представляет тиофосген, образующийся из перхлорметилмеркаптана в реакции с металлическим цинком:
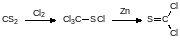
Тиофосген это реагент, с помощью которого можно получать производные тиоугольной кислоты с пестицидной и фармакологической активностью. Он представляет собой дымящую на воздухе жидкость грязно-жёлтого цвета. Тиофосген очень легко окисляется кислородом воздуха и гидролизуется влагой, т.е. его пары превращаются на воздухе в хлористый водород, серный ангидрид и диоксид углерода. Смесь этих веществ вызывает сильнейший ожог верхних дыхательных путей. При вдыхании этой смеси может произойти рефлекторная остановка дыхания – болевой эффект в носоглотке вызывает спазм её мышц, перекрывающий дыхательные пути. Следствием этого может быть летальный исход от удушья. Такой же эффект могут вызвать и другие вещества с сильным раздражающим действием на слизистые оболочки – другие хлорангидриды органических и неорганических кислот, аммиак и вещества, относящиеся к ирритантам.
Определённое синтетическое значение имеет хлорангидрид трихлорметилового эфира угольной кислоты, называемый дифосгеном. Он более безопасен в обращении, чем фосген из-за жидкой консистенции, его температура кипения составляет 127°С. Дифосген получают исчерпывающим хлорированием метилового эфира хлоругольной кислоты или метилового эфира муравьиной кислоты (его получают из муравьиной кислоты и метанола, т.е. по бесфосгенной технологии):
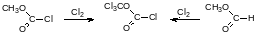
Дифосген может быть использован для получения изоцианатов. В диоксане он легко ацилирует даже соли аминов. Например, его взаимодействие с гидрохлоридом этилового эфира глицина протекает по схеме:
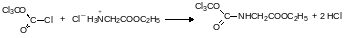
Образующееся в этой реакции соединение не выделяют в чистом виде. При нагревании оно разлагается с образованием изоцианоэтилацетата:
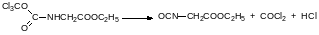
Последние десять лет большое внимание уделяется ещё одному заменителю фосгена – бистрихлорметиловому эфиру угольной кислоты с тривиальным названием трифосген. Его получают исчерпывающим хлорированием диметилкарбоната, получение которого также возможно по бесфосгенной технологии. Диметилкарбонат образуется при окислительном карбонилировании метанола под давлением в присутствии катализаторов на основе одновалентной меди:
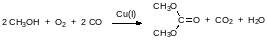
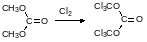
Трифосген – это кристаллическое вещество с невысоким давлением паров. Как отмечалось выше, из него можно с количественным выходом получать фосген при катализе некоторыми азотсодержащими соединениями, но в настоящее время уже разработано множество методик, позволяющих проводить реакции с его участием так, чтобы во взаимодействии участвовал образующися из трифосгена фосген в момент его образования. Так, например, непосредственно из трифосгена можно получать бензилхлорформиат, используемый для защиты аминогрупп в реакциях пептидного синтеза (Cbz‑защита). Реакция трифосгена с бензиловым спиртом в присутствии пиридина протекает по схеме:
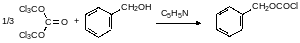
Яды крови
Для токсикологии главной компонентой крови являются эритроциты. Они предназначены живой природой для переноса кислорода клеткам организма. Их форма всем известна – это плоские круглые клетки без ядра. Такая форма нужна для того, чтобы диффузия кислорода легко проходила на небольшом расстоянии от мембраны эритроцитов. Кислород связывается с атомом железа в молекуле гема, включенной в белковую молекулу с образованием сульфидных связей в результате присоединения тиольных групп в составе белка к винильным группам в молекуле гема. Гемоглобин состоит из четырёх белковых молекул и четырёх гемов. Этим реализуется распространённый в живой природе принцип кооперативного связывания или кооперативного ответа на структурную перестройку одной из белковых молекул. Чем больше объединённых в комплекс компонент выполнило свою функцию, тем легче к ним подключаются те, которые ещё не включились. Механизм этого можно объяснить зависимостью прочности связи молекулы кислорода с гемом от кислотности среды. Кислотное окружение может генерироваться в результате перестройки глобулярной молекулы белка, но может иметь и другой источник. Так, например, у жителей высокогорий, приспособившихся к жизни в атмосфере с пониженным парциальным давлением кислорода, в крови повышено содержание дифосфоглицериновой кислоты:
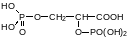
Другим фактором, определяющим кислотность в окружении гемоглобина, является диоксид углерода, образующий угольную кислоту. Повышение её концентрации в крови делает более доступным кислород оксигемоглобина.
Парциальное давление кислорода в воздухе на уровне моря составляет около 150 мм рт. ст., над артериальной кровью, выходящей из лёгких, парциальное давление в зависимости от возраста составляет от 70 до 100 мм рт. ст., а над венозной кровью – около 50 мм рт. ст. С разряжением воздуха при подъёме в высоту парциальное давление кислорода снижается и примерно на высоте 6000 м оно оказывается равным давлению кислорода над венозной кровью. Это значит, что без специальной адаптации находиться на больших высотах опасно для жизни. Альпинисты при восхождении на семи- и восьмитысячники берут кислород в баллонах.
Кровь растворяет кислород, но в связанном с гемоглобином состоянии кислорода в артериальной крови примерно в 70 раз больше. Кислород связывается с двухвалентным атомом железа в составе гема, занимая шестую его координацию. В молекуле гема четыре координации заняты атомами азота его порфириновой основы:
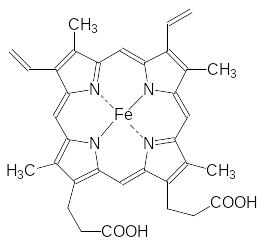
Пятая координация занята атомом азота имидазольного цикла гистидина в белковой молекуле. Пять атомов азота в окружении двухвалентного атома железа снижают его способность окисляться кислородом. Есть генетическое нарушение, при котором биосинтез белковой компоненты гемоглобина проходит с заменой представляющего для координации имидазольный цикл гистидина на тирозин. Гидроксифенильная группа тирозина координируется на атом железа, но не защищает его от окисления кислородом. Окисленный атом железа в трёхвалентном состоянии не связывается с кислородом. У такого атома железа все координации уже заняты, поскольку у шестой координации появится анион для компенсации «лишней» валентности. У людей с этим генетическим отклонением содержание гемоглобина с трёхвалентным атомом железа, его называют метгемоглобином, может достигать 50 %, тогда как в нормальном состоянии метгемоглобина в крови около 1,0 %. Понятно, что клетки организма при таком балансе гемоглобина и метгемоглобина (метгемоглобинемия) испытывают кислородное голодание. Но это не единственная проблема для здоровья. Дело в том, что одноэлектронное окисление атома железа молекулой кислорода переводит её в анион-радикал супероксид, окисляющий все органические молекулы:
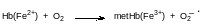
Для нейтрализации супероксида, образующегося и в нормальных условиях в эритроцитах содержатся два фермента – супероксиддисмутаза, катализирующая диспропорционирование супероксида с образованием кислорода и пероксидного анион, и каталаза, которая переводит пероксидный анион в кислород и анион воды.
Существует неферментативное восстановление метгемоглобина в гемоглобин, основанное на его восстановлении глютатионом или аскорбиновой кислотой. Однако в восстановлении метгемоглобина в гемоглобин главную роль играет фермент метгемоглобинредуктаза. Метгемоглобинредуктаза блокируется многими соединениями, например, нитробензолом и анилином, а также лекарственными препаратами, среди которых некоторые сульфамиды и противомалярийные средства.
В каталитическом центре супероксиддисмутаз находятся ионы меди, марганца и реже железа и никеля. Эти металлы переходной валентности могут переходить в некаталитическое состояние в результате восстановления. Одним из наиболее известных токсикантов, действие которого направлено на восстановительное превращение ионов металлов в ферментах, является очень токсичный газообразный арсин AsH3. Он образуется при действии кислот на арсениды металлов, например:
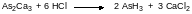
Блокировка работы супероксиддисмутазы приводит к накоплению в клетках супероксида, который окисляет ненасыщенные гидрофобные остатки в липидах клеточных мембран эритроцитов. Это переводит эти структурные элементы в гидрофильное карбоксилатное состояние. В мембранах не может больше поддерживаться их двухслойное состояние, они частично растворяются, высвобождая заключенные в цитозоле эритроцитов белки и гемоглобин. Кровь от этого сильно густеет и плохо прокачивается через кровеносные сосуды, а обрывки мембран закупоривают капилляры и почечные канальцы. Часть гемоглобина поступает в мочу, окрашивая её в красный цвет. В тяжёлых случаях отравление арсином заканчивается летальным исходом. В соответствии с этим были планы по использованию арсина в качестве боевого отравляющего вещества.
Близок по токсическим характристикам к арсину его аналог фосфин РН3. Но у него ярко проявляются и общетоксические свойства. Как арсин, так и фосфин являются сильными восстановителями. Угольный противогаз не может быть использован для защиты от этих газов, поскольку на активированном угле идёт их окисление, сопровождающееся сильным термическим эффектом. Фосфин при превышении определенной концентрации может даже поджечь шихту противогаза.
Фосфин образуется при диспропорционировании соединений фосфора с валентностью больше -3 и меньше +5. Так, например, он образуется при нагревании белого фосфора (валентность 0) в щёлочи. Диспропорционирует при нагревании фосфорноватистая и фосфористая кислота, а также неполные эфиры кислот фосфора с промежуточной его валентностью. В отличие от арсина, который не может образоваться в синтезах обычных органических соединений, с фосфином можно встретиться при работе с органическими производными кислот фосфора и в реакциях, сопровождающихся образованием фосфористой кислоты. В качестве примера можно привести получение газообразного бромистого водорода из брома и красного фосфора с добавлением в реакционную массу небольшого количества воды. Экзотермическая реакция разложения образующегося трибромида фосфора водой на бромистый водород и фосфористую кислоту сопровождается её диспропорционированием на фосфин и фосфорную кислоту. Ещё один пример представлен получением иодистоводородной кислоты без образующегося в результате её окисления иода. Для освобождения иодистоводородной кислоты от иода её перегоняют над красным фосфором. Иод реагирует с фосфором с образованием иодида состава Р2I4, разлагающегося водой с образованием иодистого водорода и кислот фосфора в низшей валентности, которые легко диспропорционируют с образованием фосфина и фосфорной кислоты. Это приводит к тому, что фосфин попадает в перегнанную иодистоводородную кислоту, образуя с ней соль.
Ещё один способ получения фосфина представлен взаимодействием фосфидов металлов с водой или с кислотами. Так, в качестве ратицида использовался фосфид цинка, образующийся при пропускании паров фосфора над расплавленным цинком или при восстановлении фосфата цинка прокаливанием с коксом:

Фосфид цинка не реагирует с водой, но в желудочном соке содержится разлагающая его соляная кислота. Сейчас использование фосфида цинка в качестве ратицида запрещено из-за отравлений птиц содержащими фосфид цинка приманками.
Есть также фосфиды металлов, разлагающиеся водой. К ним относится фосфид алюминия. Прессованные из фосфида алюминия таблетки используют для защиты складских помещений от вредителей. Выделяющийся в реакции фосфида алюминия с влагой воздуха фосфин защищает складированную продукцию от всех видов вредителей, начиная с грызунов и кончая микроорганизмами.
Очень токсичен монооксид углерода, который связывается с атомом железа в геме примерно в 250 раз прочнее кислорода из-за карбенового характера атома углерода в его составе. Монооксид углерода образуется при неполном сгорании всех видов топлива. Так, например, в выхлопных газах может содержаться до 10 % этого вещества, называемого также угарным газом. Много его в пороховых газах и в газах, образующихся при взрывах боеприпасов. Для защиты от угарного газа противогазная коробка дополняется так называемым гопкалитовым патроном, заполненным оксидом марганца, который активирован оксидами меди и серебра. Гопкалит катализирует окисление монооксида углерода до диоксида, но при низких температурах гопкалит не защищает от СО, а при длительной работе он может разогреться до высоких температур.
В лабораторных условиях монооксид углерода можно получить, прибавляя муравьиную кислоту к концентрированной серной кислоте при нагревании:
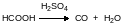
Монооксид углерода представляет собой исходный продукт в химической промышленности. Основная область его применения – это оксосинтез, разные варианты которого используются для получения многих органических соединений, в числе которых альдегиды, карбоновые кислоты, уксусный ангидрид и многие другие продукты многотоннажного синтеза. Восстановлением монооксида углерода получают метанол и углеводороды.
В качестве катализаторов при получении альдегидов в оксосинтезе используют карбонилы металлов, в частности, тетракарбонил никеля. Карбонилы металлов представляют собой в основном жидкости, образующиеся из металлов и монооксида углерода при нагревании под давлением. Наиболее известны пентакарбонил железа и тетракарбонил никеля. Его каталитическая роль в оксосинтезе описывается уравнениями:

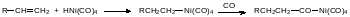
Образовавшееся после встраивания в связь атома никеля с алкильным остатком карбонильное соединение разлагается водой с образованием кислоты, а водород разрывает связь карбонильной группы с атомом никеля с регенерацией исходного гидридного соединения и альдегида:
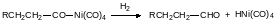
По аналогичной схеме, но с участием в реакции метилиодида при катализе соединениями родия, из метилового эфира уксусной кислоты и монооксида углерода получают ангидрид уксусной кислоты.