

§ 124. Установлення точки еквівалентності за допомогою індикаторів
Індикатори. Для визначення точки еквівалентності існують різні способи. Найбільш доступний і поширений спосіб — застосування індикаторів. Можна також ввести в розчин певний металевий електрод і в процесі титрування вимірювати його потенціал; точку еквівалентності в цьому випадку знаходять з аналізу залежності зміни потенціалу від об'єму добавленого робочого розчину. Це спосіб так званого потенціометричного титрування. Спинимось докладніше на індикаторному способі визначення точки еквівалентності.
Індикатори — це хімічні сполуки, за допомогою яких установлюють точку еквівалентності. Індикаторами можуть бути речовини, здатні вступати в хімічні реакції з реактивом А при цілком певній концентрації останнього (або з речовиною В), причому утворені продукти реакції легко помітити неозброєним оком. Продукт реакції може бути розчинною забарвленою сполукою або кольоровим чи безбарвним осадом.
Індикатори звичайно добавляють у невеликій кількості до розчину, який титрують; значно рідше користуються так званими зовнішніми індикатора-
390
ми. Класифікацію обох типів індикаторів і техніку їх застосування описано
нижче.
Треба відразу підкреслити одну важливу рису наведеного вище означення поняття індикаторів. Індикатори — це не просто які завгодно речовини, що дають з реактивом А або з речовиною В забарвлені чи малорозчинні сполуки; індикатор повинен давати помітний ефект реакції тільки при цілком певній концентрації реактиву А (або речовини В), зумовленої властивостями продукту реакції АВ. Розглянемо, наприклад, титрування хлоридів розчином нітрату ртуті (II). Визначення грунтується на реакції
Hg2++2Cl- = HgC]2.
При цій реакції утворюється малодисоційований хлорид ртуті (II). Щоб установити точку еквівалентності, до розчину хлориду треба добавити речовину, яка здатна реагувати з іонами ртуті, утворюючи помітну оком сполуку. Відомо, наприклад, що сульфід-іони реагують з іонами ртуті, утворюючи чорний осад HgS. Але іони S2- непридатні як індикатор при цьому титруванні. Перша ж краплина добавленого робочого розчину Hg (N03)2 призведе до осадження чорного сульфіду ртуті, що пояснюється дуже малою розчинністю осаду HgS. Іони ртуті в першу чергу реагуватимуть з іонами сірки, а не з іонами хлору, і точка еквівалентності буде зафіксована неправильно. Іони сульфіду в цьому випадку є надто чутливим реактивом, який фіксує присутність іонів ртуті в концентрації, значно меншій, ніж у точці еквівалентності. Для титрування хлоридів розчином Hg (N03)2 можна використати розчин нітропрусиду натрію Na2 [Fe(CN)5NO].
Останній також дає з іонами ртуті малорозчинну сполуку Hg [Fe(CN)5NO ], але, на відміну від попереднього випадку, осад Hg [Fe(CN)5NO] утворюється тільки після того, як усі іони хлору зв'язані іонами ртуті в малодисоційовану сполуку HgCl2.
З розглянутого вище прикладу випливає такий висновок: індикатор повинен реагувати з реактивом А, що застосовується для титрування (чи з визначуваною речовиною В) при такій концентрації останнього, яка має бути в точці еквівалентності.
Нижче буде показано, що при розгляді процесів титрування замість концентрації реагуючих речовин іноді зручно користуватись десятковим логарифмом числового значення концентрації, узятим з оберненим знаком. Надалі для зручності називатимемо цю величину просто від'ємним логарифмом концентрації. Позначимо від'ємний логарифм концентрації речовини А в точці еквівалентності — lg Лекв через рЛекв; від'ємний логарифм концентрації реактиву А, при якій останній вступає з реакцію з індикатором,— через р7\ Величина рГ називається показником титрування індикатора. Очевидно, умовою правильного визначення точки еквівалентності за допомогою того чи іншого індикатора є рівність цих двох величин, тобто
Р^екв = VT.
Аналогічно до цього, коли індикатор реагує з речовиною В (а не з реактивом А), найбільш сприятливою умовою точного визначення буде
РВекв = РТ-
391
Додержати рівності величин рАекп/(рВСкв) і рТ досить важко. Властивості продуктів реакції АВ в різних випадках неоднакові. Тому точне виконання цієї умови означало б потребу мати для титрування кожної речовини окремий індикатор. Звичайно в практиці нри визначенні різних речовин часто застосовують топ самий індикатор, допускаючи деяку нерівність величин рЛекв (рбекв) і рТ. Це призводить до того, що індикатор фіксує кінець тит-
РВвнв.
рт
РВ = -1дВ
|
|
|
|
|
|
|
|
, ^ґ |
/і |
а |
|
|
|
|
|
І |
|
|
|
^ п ■ 1 |
" |
|
|
—^ |
ГЇ1 |
^_- |
мл |
||
Рис. 31. Загальний вигляд кривих титрування.
рування раніше або пізніше, ніж буде досягнуто точки еквівалентності. Момент титрування, коли спостерігається зовнішній ефект реакції між індикатором і робочим розчином реактиву (чи визначуваною речовиною), називають кінцевою точкою титрування. Розбіжність між кінцевою точкою титрування і точкою еквівалентності є причиною виникнення помилки титрування; проте в більшості випадків, якщо розходження між рЛЄкв і рТ не виходить за певні межі, помилка титрування незначна і нею можна нехтувати. Важливо правильно оцінити вплив властивостей продукту реакції та інших факторів на точність титрування і вміти визначити допустиму розбіжність величин рАек„ і рТ, яка дає змогу провести титрування з достатньою точністю. З цього погляду першорядне значення має характеристика змін концентрації реагуючих речовин у процесі титрування. Розглянемо це питання докладніше.
Криві титрування і помилки титрування. Графічна залежність зміни концентрації визначуваної речовини В від об'єму робочого розчину реактиву А називається кривою титрування. При титруванні концентрації В \ А, як правило, змінюються дуже часто в мільйони і десятки мільйонів разів. Для зручності графічного позначення таких великих змін концентрації звичайно користуються логарифмічною залежністю, відкладаючи на осі ординат величину від'ємного логарифма концентрації — lg В або — lg A; на осі абсцис відкладають об'єм робочого розчину в мілілітрах. Загальний вигляд типової кривої титрування показано на рис. 31 безперервною лінією 1.
1 Наведена вище крива титрування побудована з припущенням, що об'єм розчину під час титрування не змінюється. Практично цього можна досягти, застосовуючи, наприклад,
392
Абсциса точки а позначає кількість мілілітрів робочого розчину А, витраченого на титрування до досягнення точки еквівалентності; ордината цієї точки рЯекв відповідає концентрації речовини В в точці еквівалентності.
Важлива властивість кривої титрування, як видно з рисунка, полягає в тому, що концентрація В під час титрування змінюється нерівномірно; спочатку із збільшенням об'єму робочого розчину А вона зменшується 1 мало, як видно з пологої нижньої частини кривої. У міру наближення до точки еквівалентності це зменшення стає більш різким і, нарешті, в точці еквівалентності досягає максимального значення. Після точки еквівалентності концентрація В знову змінюється повільніше (див. праву верхню частину кривої). Звідси, до речі, випливає інше визначення точки еквівалентності порівняно з наведеним вище. Можна сказати, що точка еквівалентності відповідає такому моменту титрування, коли зміна концентрації визначуваної речовини максимальна.
З рисунка ще видно, що інтервал різкої зміни концентрації В біля точки еквівалентності досить значний. Ця обставина має велике значення для правильного визначення точки еквівалентності за допомогою індикаторів. Припустимо, наприклад, що індикатор дає забарвлену сполуку з речовиною В при такій концентрації останньої, яка відповідає величині рТ (див. рис. 31) 2. Отже, на початку титрування розчин буде забарвлений; коли концентрація зменшиться до величини р7\ зміна кольору індикатора покаже на досягнення кінцевої точки титрування. У цьому випадку кінцева точка титрування настане до того, як буде досягнута точка еквівалентності. Отже, розчин буде трохи недотитрований, причому причиною цього є розходження між величинами рТ і р5екв:
Визначимо поняття помилки титрування як відношення кількості (концентрації) невідтитрованої частини речовини В в кінцевій точці титрування до її загальної кількості (концентрації) на початку титрування. Відповідно в тому випадку, коли розчин у кінцевій точці титрування пере-титрований, помилку титрування визначимо відношенням зайвої кількості введеного робочого розчину до початкової кількості речовини. У нашому прикладі невідтитровану частину можна характеризувати відрізком / на абсцисі, а загальну кількість речовини В — відрізком т. З рисунка видно, що в цьому випадку помилка незначна; це зумовлено дуже різкою зміною концентрації В поблизу точки еквівалентності. Тому навіть досить значне розходження між рТ і р5екв мало відбивається на точності титрування.
концентрований розчин реактиву або вводячи останній у сухому вигляді. Це зроблено для того, щоб мати можливість дослідити залежність —lg В від об'єму робочого розчину реактиву в чистому вигляді і виключити вплив розведення, при добавлянні робочого розчину на концентрацію В.
1 Tj?*6a пам'ятати, що на осі ординат відкладено від'ємний логарифм концентрації В, тому підйом кривої відповідає зменшенню концентрації В.
2 Загальний хід міркувань і остаточний висновок будуть такі ж самі й у випадку коли індикатор утворює забарвлену або малорозчинну сполуку з робочим розчином А.
393
Розглянемо тепер вплив зміни початкової концентрації В на форму кривої титрування і на точність останнього. Припустимо, що початкова концентрація розчину В в 10 раз менша, ніж у попередньому випадку. Тоді нижня полога частина кривої піде трохи вище, ніж раніше. Вигляд цієї кривої титрування показано на рис. 31 пунктиром. З рисунка видно, що в цьому випадку таке саме відхилення рТ від рВекв призводить до значно більшої помилки титрування, яка визначається відношенням відрізків п і гп.
рВ=НдВ
АВ
Нрива
титрування
В
реактивом
А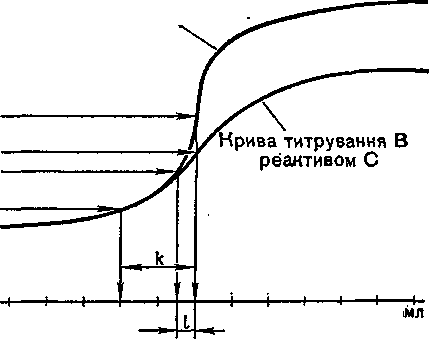
рВсв РТЛВ
св
Рис. 32. Вплив енергії хімічного споріднення між реагуючими речовинами на форму кривих і точність титрування.
Порівняємо, далі, криві титрування розчину речовини В робочими розчинами двох різних реактивів, які утворюють з В продукти реакції АВ і СВ, неоднакові за своїми властивостями. Нехай енергія хімічного споріднення між А і В більша, ніж енергія споріднення між С і В. Отже, зворотна реакція з утворенням вихідних продуктів заходить у випадку СВ далі, ніж у випадку А В. Це означає, що концентрація В в точці еквівалентності при титруванні розчином реактиву С значно більша, ніж при титруванні розчином реактиву А. Відповідно до цього криві титрування мають вигляд, показаний на рис. 32.
На початку титрування обидві криві збігаються. Поблизу точки еквівалентності крива С має менший нахил, ніж крива А, а після точки еквівалентності полога частина кривої С проходить нижче від пологої частини кривої А. Концентрація В в обох точках еквівалентності позначена на рисунку відповідно ординатами рВлв і рВСв- Припустимо, що в обох випадках застосовують індикатори, показники титрування яких рТлв і рТсв відрізняються від відповідних величин рВлв і рВсв на ту саму величину, як це видно з рис. 32:
VBAB — VTAB = VBCB — VTCB-
Ш
З рисунка видно, що помилка визначення В при титруванні реактивом С буде значно більшою порівняно з помилкою, яка спостерігається при титруванні реактивом А. Обидві помилки характеризуються відрізками / і k.
З розглянутих вище положень випливають такі загальні висновки. Помилка титрування збільшується із збільшенням розходження між величинами рВек,. і рТ. При даній величині розходження між показником титрування індикатора рГ і концентрацією речовини В в точці еквівалентності помилка титрування зростає із зменшенням початкової концентрації В і ослабленням енергії хімічного споріднення між В та реактивом, розчин якого застосовують при титруванні. Точність титрування розведених розчинів завжди менша, ніж концентрованих, тому в титриметричному аналізі користуються звичайно 0,1 н. розчинами.
Точність титрування збільшується також із зменшенням швидкості зворотної реакції або зростанням споріднення між А і В. Тому при титруванні, наприклад, методом осадження треба вибирати такі робочі розчини, які при взаємодії з визначуваною речовиною давали б найменш розчинні сполуки. Так, титрування іонів срібла іонами йодиду дає точніші результати, ніж титрування іонами хлориду. Це пояснюється меншою розчинністю осаду Agl порівняно з осадом AgCl. Титрування методом комплексоутво-рення чи кислотно-основне титрування дає тим кращі наслідки, чим менша дисоціація чи гідроліз продуктів реакції. У цих випадках інтервал різкої зміни концентрації речовини В поблизу точки еквівалентності буде досить значним (рис. 32). Тому навіть помітна розбіжність між величинами р5еКь і рТ не призведе до великої помилки титрування. Конкретні приклади обчислення помилки титрування і вибору найпридатніших індикаторів наведені в наступних розділах, де розглядаються відповідні методи титриметрич-ного аналізу.
Класифікація індикаторів. Індикатори поділяються на окремі групи за різними принципами. За технікою застосування розрізняють внутрішні і зовнішні індикатори. Внутрішні індикатори — це речовини, що вводяться для встановлення точки еквівалентності безпосередньо в розчин, який титрують. Такі індикатори найзручніші, через що вони дуже поширені. Кінцеву точку титрування розпізнають за зміною кольору індикатора в розчині або за появою чи зникненням осаду. Проте бувають окремі випадки, коли внутрішніми індикаторами користуватись незручно або неможливо. Це буває, наприклад, тоді, коли при титруванні утворюються забарвлені осади, що маскують зміну кольору індикатора. Наприклад, іони нікелю можна визначити за допомогою титрування розчином диметилгліоксиму. Тут внутрішні індикатори непридатні, тому що осад диметилгліоксимату нікелю забарвлений у червоний колір. Визначення з зовнішнім індикатором ведуть, відбираючи в процесі титрування час від часу краплину розчину і встановлюючи за допомогою якісної реакції присутність у розчині іонів нікелю. Перше титрування звичайно вважають орієнтовним. При повторному титруванні, коли вже приблизно відома кількість робочого розчину, що витрачається на визначення, добавляють одразу майже всю цю кількість робочого розчину і роблять краплинні проби на іони нікелю тільки поблизу точки
395
еквівалентності. Для титрування із зовнішніми індикаторами треба мати досить великий практичний досвід і затрачати значно більше часу, ніж для титрування з внутрішніми індикаторами. Тому такі визначення застосовують порівняно рідко.
Оборотні і необоротні індикатори. Оборотні індикатори — це сполуки, які здатні існувати в двох формах, причому реакція переходу однієї форми в іншу і навпаки є оборотною. До цього типу належить більшість відомих індикаторів. Забарвлення оборотних індикаторів легко змінюється в обох напрямах: певному надлишку реактиву А відповідає одна забарвлена форма індикатора; навпаки, введення в розчин надлишку речовини В зумовлює перехід цієї форми в іншу і призводить до зміни кольору або його зникнення. Прикладом оборотних індикаторів є метилоранж. У лужному середовищі він забарвлений у жовтий колір, а в кислому — в червоний. Зміна червоного забарвлення на жовте або навпаки може відбуватися в обох напрямах багато разів залежно від присутності в розчині надлишку кислоти або лугу. Оборотні індикатори зручні й тим, що при випадковому введенні надто великої кількості робочого розчину визначення все-таки можна закінчити правильно: для цього до перетитрованого розчину треба добавити ще певну кількість визначуваної речовини (при цьому індикатор відновить свій попередній колір) і обережно дотитрувати розчин.
У деяких випадках доводиться користуватись необоротними індикаторами. Необоротні індикатори — це такі сполуки, які під впливом надлишку реактиву руйнуються і попереднє забарвлення яких не відновлюється від добавляння надлишку визначуваної речовини. Прикладом необоротних індикаторів може бути той самий метилоранж, якщо він застосовується при титруванні розчином бромату калію. Робочий розчин бромату калію використовують при визначенні тривалентних миш'яку, сурми і деяких інших відновників. До того часу, поки в розчині є іони миш'яку чи сурми, бромат калію витрачається на їх окислення і не реагує (або майже не реагує) з метилоранжем. Після досягнення точки еквівалентності бромат калію вступає в реакцію з метилоранжем, причому відбувається необоротне окислення метилоранжу. Недоліком необоротних індикаторів є те, що їх забарвлення часто слабшає і навіть повністю зникає ще до точки еквівалентності. Це спостерігається в тих випадках, коли розчин погано перемішують, і пояснюється місцевим надлишком реактиву, який утворюється під час титрування в окремих місцях розчину. Тому в процесі титрування часто доводиться добавляти ще нові порції індикатора, щоб пересвідчитись в тому, що забарвлення індикатора зникло внаслідок введення надлишку робочого розчину, а не внаслідок поступового окислення індикатора під час добавляння робочого розчину.
Індикатори розрізняють ще за типом хімічної реакції, що лежить в основі визначення. Відповідно до цього існує кілька груп індикаторів.
Індикатори методів кислотно-основного титрування. Це забарвлені речовини органічного походження, які здатні існувати в двох формах, залежно від величини рН розчину. Вони належать до групи оборотних індикаторів. Найчастіше обидві форми цих індикаторів забарвлені в різний колір;
396
це так звані двоколірні індикатори. Рідше використовуються індикатори одноколірні, тобто такі, в яких забарвлена тільки одна форма. Кислотно-основні індикатори реагують на зміну концентрації водневих іонів і в цьому розумінні є специфічними індикаторами на водневі
іони.
Індикатори методів окислення-відновлення. Більшість цих індикаторів є також органічними речовинами, що можуть існувати в двох формах — окисленій і відновленій. Але, на відміну від кислотно-основних індикаторів, вони не специфічні відносно певних іонів; забарвлення змінюється, як правило, залежно від зміни величини окислювально-відновного потенціалу розчину. Майже всі індикатори методів окислення-відновлення є оборотними (див. §135).
Індикатори методів осадження. Ці індикатори здебільшого характеризуються специфічною дією відносно певних іонів. У цій групі слід окремо виділити так звані адсорбційні індикатори (див. §147).