

Учебник Органическая химия Щеголев 2016
.pdfметилфосфоновая кислота диметилфосфиновая кислота триметилфосфиноксид
Фосфины, за исключением газообразного метилфосфина, представляют собой бесцветные жидкости или низкоплавкие твѐрдые вещества с неприятным запахом. Они нерастворимы в воде и чрезвычайно ядовиты. Молекулы триалкилфосфинов имеют пирамидальную конфигурацию.
По свойствам они похожи на амины. Обладают оснóвными свойствами (для триметилфосфина рКb = 5.3) — при взаимодействии с кислотами образуют соли замещѐнного фосфония:
R3P + H+ R3PH+
Фосфины легко алкилируются с образованием солей фосфония, окисляются кислородом или другими окислителями, например, пероксикислота-
ми R-COOOH (гл. 6.4.6):
|
|
|
|
|
|
|
|
R |
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
R'-X |
|
R |
P |
R' |
X- |
|||
|
|
|
|
|
|
|
||||||
R |
|
|
||||||||||
|
|
|
|
|
|
|
|
|
||||
R P |
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
|||
|
|
|
|
|
|
|
|
|||||
R |
[O] |
|
|
|
|
|
||||||
|
|
|
|
|
R P |
|
|
|
|
|||
|
|
|
|
|
|
|
|
O |
||||
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|||||
R
Исходными веществами для получения фосфинов являются трихлорид фосфора PCl3 и металлорганические соединения:
PCl3 |
RMgCl |
RPCl2 |
RMgCl |
R2PCl |
RMgCl |
R3P |
||||
|
|
|
|
|
|
|||||
- MgCl2 |
- MgCl |
- MgCl |
||||||||
|
|
|
|
|||||||
|
|
|
|
2 |
|
2 |
|
|||
Фосфониевые соли в целом аналогичны солям замещѐнного аммония. Они представляют собой белые кристаллические вещества, хорошо растворимые в воде. Большинство солей первичного и вторичного фосфония при комнатной температуре неустойчиво; в растворах существует равновесие, например:
[RPH3]+X- 
 RPH2 + HX
RPH2 + HX
Наиболее устойчивы четвертичные соли, многие из которых разлагаются при температурах выше 200 С.
Своеобразно поведение фосфониевых солей, содержащих -водо- родный атом, при действии на них литийорганических соединений — образуются алкилиденфосфораны:
181
+ |
|
|
- |
|
|
|
|
|
|
||
R3P-CH-R' |
X |
- |
|
R3P=CH-R' + R"H + LiX |
|
|
|
+ R"-Li |
|
||
|
|
||||
H |
|
|
|
|
|
Фосфониевые соли легко образуются, как было указано выше, при алкилировании фосфинов.
Фосфиноксиды — это обычно бесцветные жидкости без запаха, малорастворимы в воде. Пространственное строение близко к тетраэдрическому. Это стабильные соединения, не гидролизуются, трудно восстанавливаются до фосфинов. Являются слабыми основаниями:
R3P=О + H+ R3P+-ОH
Фосфиноксиды легко образуют комплексы с ионами различных тяжѐлых металлов, поэтому применяются в качестве экстрагентов для выделения из растворов лантаноидов и актиноидов:
R |
|
R |
|
|
|
R |
|||||
2 R P |
|
O + Mn+ |
|
R P |
|
O Mn+ |
O |
|
P |
|
R |
|
|
|
|
||||||||
|
|
|
|
||||||||
|
|
|
|
||||||||
R |
|
R |
|
|
R |
||||||
Фосфиноксиды получают окислением из фосфинов (см. выше) или алкилированием оксохлорида фосфора POCl3 металлорганическими соединениями:
POCl3 RMgCl
- MgCl2
Cl
R P O RMgCl
Cl - MgCl2
R |
|
RMgC |
l R3P=O |
|
R P |
O |
|||
|
||||
Cl |
|
- MgCl2 |
||
|
|
|
||
Фосфонистые и фосфинистые кислоты являются бесцветными жид-
костями или кристаллическими веществами; умеренно растворимы в воде, легко растворяются в щелочах с образованием солей.
Эти соединения относятся к слабым кислотам. Фосфонистые и фосфинистые кислоты существуют в двух таутомерных формах, которые переходят друг в друга через образование аниона:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
OH |
O |
+ H+ |
|
|
O |
||||||||||||
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
R P OH |
R P OH |
|
|
|
R P OH |
||||||||||||
|
|
|
|
|
|
|
|||||||||||
алкилфосфонистая кислота |
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
OH |
O |
+ H+ |
|
|
|
|
|
|
O |
||||||||
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
R P R |
R P R |
|
|
|
R P R |
||||||||||||
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
182 |
|
|
|
|
|
|
|
|
|
||

диалкилфосфинистая кислота
Получают фосфонистые и фосфинистые кислоты из алкилхлорфосфинов гидролизом:
RPCl2 + 2 H2O RP(OH)2
R2PCl + H2O R2P-OH
Фосфоновые и фосфиновые кислоты — бесцветные жидкости или кристаллические вещества; растворимы в воде.
Фосфоновые, а также фосфиновые кислоты являются сильными кислотами (для фосфоновых кислот рКа1 2 2.5, а для фосфиновых — рКа 3). Они образуются при окислении фосфонистых и фосфинистых кислот.
Распространѐнным способом получения является частичное алкилирование оксохлорида фосфора и последующий гидролиз полупродуктов:
|
|
RLi |
Cl |
|
|
|
RLi |
|
|
R |
|
|
H2O |
R |
|
|
POCl3 |
|
R P |
O |
|
|
|
R P O |
|
|
R P |
O |
|||||
|
- LiCl |
|
- LiCl |
|
- HCl |
|||||||||||
|
|
Cl |
|
|
|
Cl |
HO |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
2H2O |
HO |
|
диалкилфосфиновая кислота |
||||||||
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
R |
P |
O |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
- 2HCl |
|
|
|
|
|
|
|||||||
|
|
|
|
HO |
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
алкилфосфоновая кислота
Производные фосфоновых кислот (сложные эфиры, тиоэфиры, амиды) используются в качестве пестицидов, лекарственных веществ, экстрагентов и боевых отравляющих веществ.
Как лекарственные вещества отдельные представители эфиров фосфоновых кислот применялись в качестве миотических средств для снижения внутриглазного давления при глаукоме (фосфакол, пирофос, армин, хлорофтальм и др.). Однако они вызвали слишком длительный и сильный миоз, нарушение аккомодации, оказывали раздражающее действие. В номенклатуре лекарственных средств в настоящее время сохранился лишь армин, являющийся эфиром этилфосфоновой кислоты.
|
|
|
O |
|
NO2 |
||
|
|
|
|
||||
C H |
|
P O |
|
|
|||
|
|
||||||
2 |
5 |
|
|
|
|
|
|
|
|
|
OC2H5 |
|
|
||
|
|
|
|
|
|||
|
|
|
армин |
|
|||
183

Из боевых отравляющих веществ известны зарин и зоман. Это вещества, обладающие нервно-паралитическим действием, вызывают поражение при любом способе воздействия, особенно быстро — при ингаляции.
|
|
O |
|
|
|
CH3 |
|
|
O |
|
|
CH3 CH3 |
||||||||
H3C |
|
P |
|
O |
|
CH |
H3C |
|
P |
|
O |
|
CH |
|
C |
|
CH3 |
|||
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
CH3 |
|
|
|
||||||||||
|
F |
|
|
|
|
F |
|
|
|
|
CH3 |
|||||||||
|
|
|
|
|
зарин |
|
|
|
|
|
зоман |
|||||||||
3.9.3. МЫШЬЯКОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Мышьяк, подобно фосфору, в составе органических веществ образует несколько групп соединений. Это арсины, арсиноксиды, арсиновые кислоты и др. Закономерности в изменениях свойств этих соединений обусловлены сходством и различием природы атома мышьяка по сравнению с фосфором, находящихся в одной группе Периодической таблицы.
Арсины очень ядовиты; некоторые из них применялись в качестве боевых отравляющих веществ. Среди них наиболее ядовитым является люизит.
Cl
Cl As CH=CHCl
люизит ( -хлорвинилхлорарсин)
Люизит — маслянистая жидкость, в малых концентрациях обладает запахом, напоминающим запах герани. Он относится к группе отравляющих веществ кожно-нарывного и общетоксического действия.
Арсиновые кислоты аналогичны фосфиновым кислотам, например:
CH3 CH3 As O
HO
диметиларсиновая кислота (какодиловая кислота)
Арсиновые кислоты гораздо менее ядовиты, чем арсины. На их основе была создана группа лекарственных препаратов для лечения сифилиса, а натриевая соль какодиловой кислоты применялась в дерматологии.
Однако в настоящее время препараты мышьяка применяют редко. В номенклатуре лекарственных средств остался лишь осарсол (в составе препарата осарбон), относящийся к арсоновым кислотам:
184
|
|
|
|
|
|
OH |
||
|
|
|
|
|
|
|||
HO |
|
|
As |
|
O |
|||
|
|
|
||||||
|
|
|
|
|
||||
O |
|
OH |
||||||
|
||||||||
|
|
|
|
|
||||
H3C-C-HN |
|
|
|
|||||
|
|
осарсол |
|
|
|
|||
и такие производные арсенобензола, как новарсенол и миарсенол.
HO |
As As |
OH |
H2N |
|
NH-CH2-SO2Na |
|
новарсенол |
|
HO |
As As |
OH |
NaO3S-CH2-HN |
|
NH-CH2-SO3Na |
|
миарсенол |
|
Однако исторически первым наиболее известным органическим соединением мышьяка была «окись какодила» – тетраметилдиарсиноксид. Это вещество было открыто в 1760 году при нагревании оксида мышьяка (III) с ацетатом калия:
|
|
|
|
H C |
|
CH3 |
|
As2O3 |
+ 4 CH3COOK |
|
3 |
As-O-As |
|
+ 2 K2CO3 + 2 CO2 |
|
|
|
H3C |
CH3 |
||||
|
|
|
|
|
|
||
тетраметилдиарсиноксид («окись какодила»)
Реакция может служить для открытия уксусной кислоты (гл. 6.4.7): сильный, отвратительный запах тетраметиларсиноксида делает еѐ чрезвычайно чувствительной.
ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите по заместительной номенклатуре ИЮПАК:
185

CH3 - |
|
CH - Br |
CH3 |
|
|
|
|
OH |
CH3 |
|
||
|
|
|||||||||||
CH3 - CH - CH2Cl |
HO - C - CH2 - C - CH3 |
CH3 - C - NH2 |
|
|||||||||
|
|
|
|
|
CH3 |
|
|
|
|
CH3 |
CH3 |
|
BrCH2 - CH2 - |
|
CF - CH2Br HOCH2 - |
|
CH - OC2H5 |
CH2CH2CH3 |
|
||||||
|
|
|
||||||||||
|
|
CH3 - CH - CH3 |
|
CH |
CH3 - N |
|
||||||
|
|
|
|
|
|
3 |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
CH2CH3 |
|
CBr3 - CFCl2 |
CH3 - |
|
CH - OH |
CH3 |
|
|||||||
|
|
|||||||||||
|
|
|
|
|
CH3O - CH - OH |
N - CH2 - NH - CH3 |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
CH3 |
. |
Какие из приведѐнных соединений могут быть названы по радикалофункциональной номенклатуре, по «а»-номенклатуре? Дайте эти названия.
2.Изобразите проекции Фишера: а) (S)-2-метилбутан-1-ол, б) (2R,3R)-2- бром-3-хлорпентан.
3.Изобразите проекции Ньюмена наиболее стабильных конформаций следующих соединений: а) (2R,3S)-бутан-2,3-диол, б) (2S,3S)-бутан-2,3-диол.
4.Сравните кислотные свойства следующих спиртов: метанол; пропан-2- ол; бутан-2-ол; неопентиловый спирт; 2,2,3,4,4-пентаметилпентан-3-ол; бутан-2,3-диол; бутан-1,4-диол; глицерин.
5.Сравните оснόвные свойства следующих аминов: изобутиламин; бутан-1- амин; 2-метилбутан-2-амин; этан-1,2-диамин; 2-метилпропан-1,2-диамин; диметиламин; метилэтиламин; диэтиламин; диметилэтиламин; трипропиламин.
6.Какие из органических реакций наиболее типичны и являются общими для галогеналканов, спиртов, простых эфиров и аминов? Охарактеризуйте реакции нуклеофильного замещения и отщепления. По каким механизмам могут протекать эти реакции? От чего это зависит?
7.Приведите наиболее вероятные механизмы реакций взаимодействия каждого из соединений: неопентилбромида, трет-амилбромида, изоамилбромида, 3-метил-3-бромпентана с водным раствором КОН.
8.Для н-пропилового спирта приведите механизм реакции образования в присутствии концентрированной серной кислоты простого эфира (симметричного).
9.Получите по реакции Вильямсона втор-бутил-трет-бутиловый эфир. Приведите механизм реакции. Почему для этой цели нельзя использовать трет-бутилгалогенид и втор-бутилат натрия?
10.Приведите наиболее вероятные механизмы реакций взаимодействия каждого из соединений: 1-бром-2-метилбутана и 2-бром-2-метилбутана с твѐрдым гидроксидом натрия.
186
11.Какой из изомерных хлорпентанов при дегидрохлорировании региоселективно превращается в пент-2-ен? Приведите наиболее вероятный механизм этой реакции.
12.Для изоамилового спирта приведите механизм реакций образования алкена в присутствии концентрированной серной кислоты.
13.Приведите механизмы реакций внутримолекулярной дегидратации следующих спиртов: а) 2-метилпентан-3-ола, б) 2,3-диметилбутан-2-ола, в) 3-метилбутан-1-ола, г) 2,2-диметилпентан-1-ола. Приведите все возможные направления и укажите преимущественное в зависимости от выбранных условий.
14.Приведите механизмы реакций взаимодействия с азотистой кислотой следующих аминов: метилэтиламин, метиламин, трет-бутиламин, метилэтилпропиламин.
187
Глава 4. НЕПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ
Непредельные, или ненасыщенные, углеводороды содержат в своѐм составе кратные углерод-углеродные связи, то есть двойные и тройные связи между атомами углерода. Это значит, что помимо -связей в молекулах имеются -связи между атомами углерода. Такими углеводородами являются этиленовые, содержащие одну двойную связь >C=C< ; ацетиленовые — с одной тройной связью -С С- ; а также углеводороды с несколькими кратными связями, среди которых наиболее распространены диеновые — с двумя двойными связями.
4.1. ОБЩНОСТЬ СТРОЕНИЯ И ХИМИЧЕСКИХ СВОЙСТВ
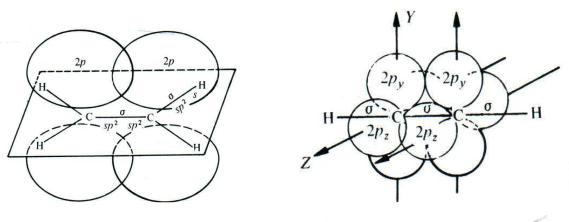
Образование -связи между атомами углерода в молекулах непредельных углеводородов происходит за счѐт бокового перекрывания негибридных р-орбиталей. Оси этих р-орбиталей перпендикулярны осям -связей. При этом в молекуле этилена между атомами углерода имеется одна -связь, а в молекуле ацетилена — две -связи (за счѐт перекрывания двух пар р- орбиталей):
этилен |
ацетилен |
В молекуле этилена все пять -связей (одна связь C–C и четыре связи C–Н) образованы sp2-гибридными орбиталями (гл. 1.2), оси которых лежат в одной плоскости. Поэтому и все шесть атомов, образующих молекулу этилена (два атома углерода и четыре атома водорода), тоже лежат в одной плоскости. Максимум электронной плотности -связи находится по обе стороны этой плоскости. Длина C=C-связи в этилене меньше, чем длина C–C-связи в этане (табл. 1.1), так как sp2-гибридные орбитали, за счѐт которых происходит образование -связей, менее протяжѐнны, чем sp3.
188

В молекуле ацетилена все три -связи (одна связь C–C и две связи C–Н) образованы sp-гибридными орбиталями, оси которых расположены под углом 180 , то есть лежат на одной прямой. Поэтому и все четыре атома молекулы (два атома углерода и два атома водорода) также располагаются на одной прямой. Максимум электронной плотности обеих -связей находится вокруг этой прямой. Длина C C-связи в ацетилене ещѐ меньше, чем длина углерод-углеродной связи в этилене (табл. 1.1), так как sp-гибридные орбитали, за счѐт которых происходит образование -связей, менее протяжѐнны, чем sp2.
Таким образом, атомы углерода, образующие -связи, в молекулах непредельных углеводородов экранированы -электронной плотностью. Это препятствует взаимодействию этих атомов с нуклеофильными реагентами, но становится возможной электрофильная атака (за счѐт электростатического притяжения -электронной плотностью углерод-углеродной связи катиона электрофила или положительного полюса молекулы, проявляющей электрофильные свойства) и, как мы увидим позднее, атака радикальных частиц. Так как прочность -связи меньше, чем -связи, то при взаимодействии непредельного углеводорода с атакующей частицей в первую очередь должна разрываться именно -связь, что приведѐт к образованию новых -связей атомов углерода с частицами реагента:
R |
R' |
|
R |
R |
|||
|
+ X-Y |
|
|
|
|
|
|
C = C |
|
|
R" - C - C - R"' |
||||
|
|
||||||
R" |
R"' |
|
|
|
|
|
|
|
X |
Y |
|||||
Возможность реакции с электрофильными и радикальными частицами с образованием продуктов присоединения может означать, что для данных соединений должны быть характерны реакции электрофильного и радикального присоединения (AE , AR).
4.1.1. РЕАКЦИИ ЭЛЕКТРОФИЛЬНОГО ПРИСОЕДИНЕНИЯ
Первой стадией электрофильного присоединения (AE) является атака электрофильной частицей субстрата, содержащего кратную связь. Пара электронов -связи более диффузна, слабее удерживается ядрами атомов углерода и поэтому является хорошим объектом для атаки электрофилом. Первоначально образуется -комплекс, затем происходит гетеролитическое расщепление -связи и образование новой -связи (C–Y):
189
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
R' |
|
|
|
R |
X - Y R' |
|
R |
X |
+ Y - |
Y X |
||||||||
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|||||
|
C = C |
+ X - Y |
|
C = C |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
C - C - R' |
|
|
R - C - C - R' |
||||||||||
|
|
|
|
|
|
R" |
- Y |
- |
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
R" |
R"' |
|
|
|
|
|
|
R"' |
|
R" |
R"' |
|
|
R" R"' |
|||||
|
|
|
|
|
|
|
-комплекс |
|
|
|
-комплекс |
|
|
|
|
|
|||
При этом атакующая частица должна быть либо катионом, либо положительной частью диполя. На последней стадии образующийся карбокатион соединяется с частицей, несущей неподелѐнную пару электронов, обычно это ион (Y ). Установлено, что в ряде случаев (при присоединении галогенов, например) образующийся карбокатион имеет мостиковую структуру:
X
C

 + C
+ C
В этих случаях атака нуклеофила на заключительной стадии осуществляется со стороны, противоположной вступлению электрофила.
Разрыв -связи, то есть превращение -комплекса в карбокатион, является наиболее медленной, лимитирующей стадией этого процесса, поэтому скорость реакции в целом будет зависеть от концентрации и субстрата, и электрофила.
Стереохимия электрофильного присоединения бывает различной. В случаях существования интермедиата (промежуточного соединения) исключительно в виде мостикового иона подход нуклеофила может осуществляться только с противоположной стороны плоскости молекулы субстрата, что приводит к анти-присоединению:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
X - Y |
|
|
X |
|
|
|
|
X |
||
|
|
|
|
|
|
|
|
|
|
|
- |
|
|||||
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
+ |
+ Y |
|
|
|
||||||
|
+ X - Y |
|
|
|
|
|
|
|
|
||||||||
C = C |
C = C |
|
|
C C |
|
|
|
- C - C - |
|||||||||
|
|
|
|
|
|
- Y - |
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Y |
||
Но для реакций, идущих без образования мостикового иона, предсказать стереохимию не так легко. Если этот ион имеет относительно большое время жизни, то нуклеофил будет атаковать с любой стороны. Если же существует какой-либо фактор, стабилизирующий определѐнную конфигурацию катиона, то тогда нуклеофильная частица будет присоединяться либо с той же, либо с противоположной стороны, в зависимости от конфигурации.
190